










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



The COVID-19 vaccine is just one example of the rapid and global effort to stopping the pandemic. Drugs too are being developed. A new study by CiRA researchers shows that the combination of two drugs halts the infection of SARS-CoV-2, the virus responsible for COVID-19, in iPS cells.
The influence of the COVID-19 pandemic can be seen in almost all industries. Chain supplies are suffering worldwide, and companies everywhere are adjusting to having their employees work from home. Science has been affected in many ways too.
CiRA as a whole has diverted massive resources to the problem, because iPS cells offer an attractive model to study the infection for several reasons.
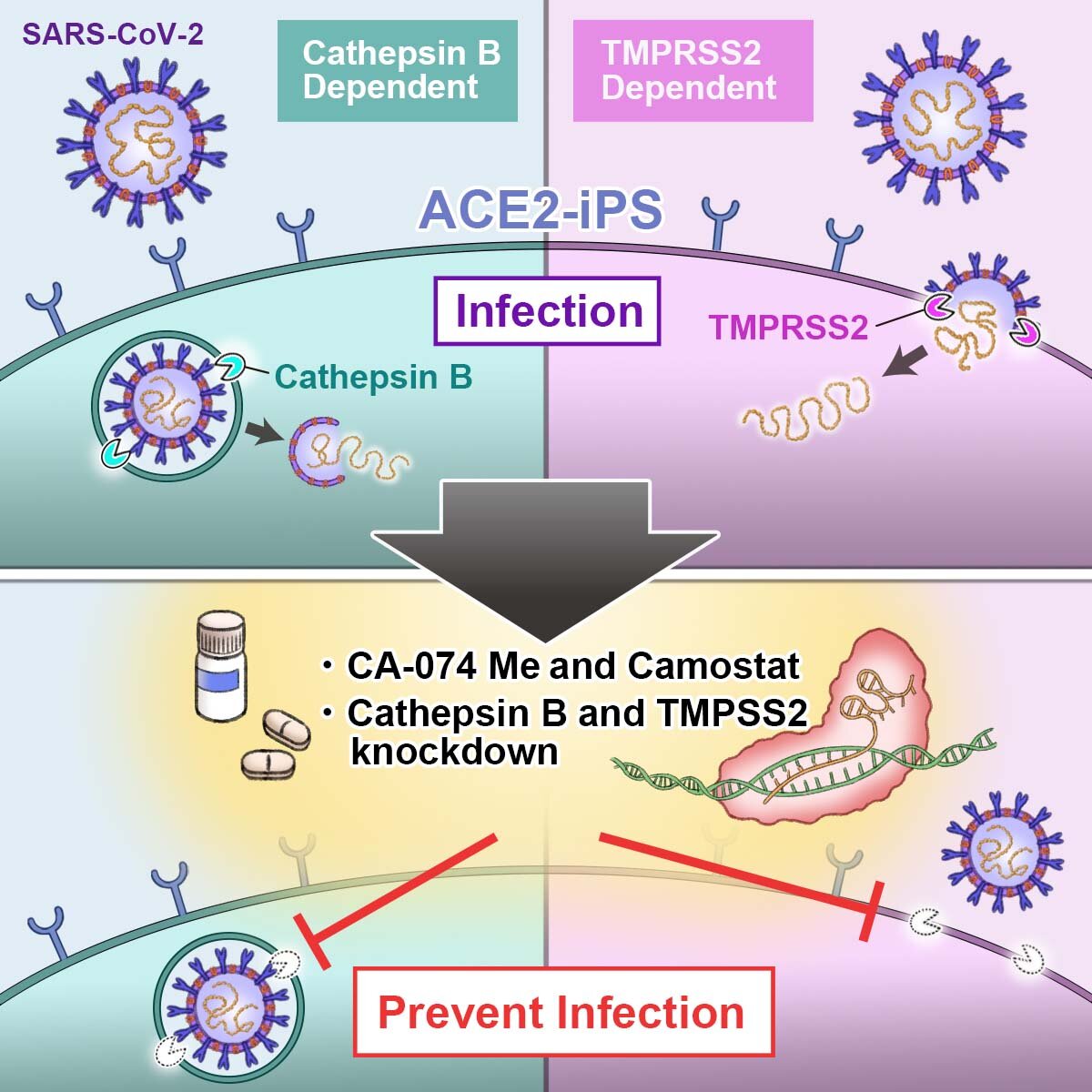
“We can differentiate iPS cells into any cell type we want to study. We can acquire iPS cells from mild and severe COVID-19 patients,” said CiRA Junior Associate Professor Kazuo Takayama about some of the benefits of these cells.
In the study, these two genes were edited so that the cell did not produce the proteases. This significantly attenuated the infection of iPS cells by SARS-CoV-2.
Using this information, the researchers tested two drugs, CA-074, which inhibits cathepsin B, and Camostat, which inhibits transmembrane serine protease 2, showing their combination reduced the viral load to less than 0.01% of that without drug treatment.
This synergy could reflect the different locations of the proteases in the cell. Cathespin B is found in endosomes, while TMPRSS2 is found in the cell membrane. Endosomes are molecules to be transported from one location to another in a cell, while proteins in the membrane can move around but only on the membrane and exchange items from the inside and outside of the cell.
However, Takayama cautions that iPS cells do not exist in patients and that more study is needed to confirm the effect of the drug combination in patient care.
“We need to see if the same effects are found in differentiated iPS cells,” he said.
Targeting Host Protease to Block Viral Entry
The proteolytic cleavage of S protein at the S1/S2 and S2′ sites by the serine protease TMPRRS2 and/or endosomal cysteine proteases CatB/L drives viral entry through the fusion peptide that inserts into the host cell membrane. This insertion leads to the formation of an antiparallel six-helix bundle, allowing the fusion process and, therefore, the uncoating and release of the viral RNA in the cytoplasm.
The S1/S2 and S2′ priming events by host proteases are necessary for SARS-CoV-2 to infect the host and interfering with the virus entry may turn out to be an advantageous antiviral strategy, as it would block the infection or virus propagation at an early stage, more importantly if considering its high transmissibility. Targeting host factors has the advantage of reducing the possible development of drug resistance and of likely providing broad-spectrum activity, by contrast interacting with the host protein, the possibility to have more severe side effects are higher with respect the classical antiviral approach.
However, TMPRRS2 and CatB/L involvement in viral infection is still the object of study and targeting these host proteases for CoV treatment is an emerging strategy. In addition, the available data suggest that simultaneous inhibition of both proteases is required for robust block of antiviral entry [58,59].
So far, there are no reports of medicinal chemistry programmes focusing of TMPRRS2 and cathepsin B/L within CoV drug discovery, thus available inhibitors often show limitations, in term of potency, selectivity, drug-like properties. Nevertheless, some compounds have been shown to exert a promising antiviral effect against SARS-CoV-2 and/or other related CoVs and are described below.
TMPRSS2 as Host Target and Its Inhibitors
Recently, TMPRSS2 has been shown to mediate SARS-CoV-2 S protein priming, as well as for SARS-CoV and other CoVs [21,58,60,61,62]. TMPRSS2, also named Epitheliasin, is a 492 aa serine protease of type II transmembrane serine proteases (TTSPs) family, expressed on the cell surface, consistently to their role in regulating cell-cell and cell-matrix interactions. The human TTSP family includes 17 members so far sharing the same structural features.
The N-terminal intracellular domain preserves the phosphorylation sites, followed by the transmembrane domain, and the stem region that is located in the initial extracellular part with a binding site for low-density lipoprotein (LDL) and calcium in a LDL Receptor A motif and a single scavenger receptor Cys-rich (SRCR domain). The C-terminal extracellular endoprotease domain contains the catalytic triad composed by the residues His-Asp-Ser, in which the Ser hydroxyl group promotes nucleophilic attack on the priming site (Figure 10) [63].
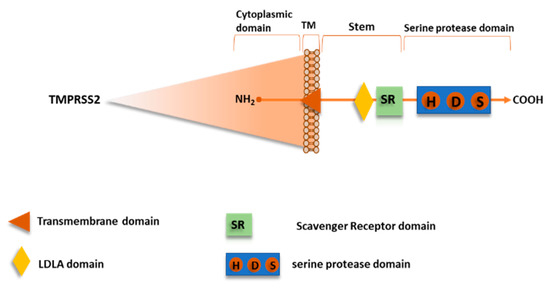
TMPRSS2 is predominantly expressed in prostate, but it has also been found in lungs, colon, liver, kidneys, and pancreas. The expression in the upper airways, bronchi and lungs, where its physiological function remains unclear, gives thought to its important role for pneumotropism of several highly pathogenic viruses, such as SARS-CoV-2, SARS-CoV, MERS-CoV, and HCoV-NL63.
Indeed, CoVs engage ACE2 to enter into host cells and although the ACE2 is a ubiquitous enzyme, they show particular tropism for the lungs. In addition, while ACE2 is expressed in both type I and type II pneumocytes, it has been verified that SARS-CoV readily infects the type I pneumocytes at early stage [60,64]. In vivo experiments showed that TMPRSS2 is responsible of viral spread and immunopathology of CoVs infection [59].
In TMPRSS2_ko mice, SARS-CoV and MERS-CoV showed significantly reduced viral replication in the lungs, especially in the bronchioles, and the inflammatory infiltration. Moreover, TMPRSS2 is not only involved with CoVs S protein activation, but its role has also been recognized in the activation of glycoprotein on the surface of influenza A virus, metapneumovirus, and porcine epidemic diarrhoea virus in different stages of viral life cycles [65].
Therefore, targeting TMPRSS2 could be a broad-spectrum antiviral strategy; however, neither drugs able to specifically inhibit this target have been identified, nor sufficient information about the substrate specificity and no 3D structures of the protein are available. However, it was reported that the fluorogenic trypsin substrates Cbz-Gly-Gly-Arg-AMC [66] and Boc-Leu-Gly-Arg-AMC [67] were also substrates for TMPRSS2, indicating that P1 can be represented by Arg, which is consistent with the recognition elements of some drugs able to inhibit human epithelia serine proteases; anyway enzymatic kinetics analyses were not performed and values of Km or Kcat are not known [67]. On the other hand, drugs that are able to inhibit a wide panel of human serine proteases, including TMPRSS2, are currently approved to treat prostate cancers and several inflammatory pathologies [68].
Previous evidences showed that the clinically proven serine protease inhibitor camostat mesylate was able, even at high concentration (up to 100 µM), to partially block SARS-CoV infection (65% of inhibition) in cell expressing TMPRSS2 without causing toxicity; hence, the antiviral activity was enhanced by adding a CatB/L inhibitor, namely E64d, thus indicating that the remaining 35% was attributable to endosomal cathepsins [58].
Camostat is a pseudo irreversible inhibitor of different serine protease, including TMPRSS2, being characterized by aromatic guanidine as P1 mimetic recognition elements (Figure 11), less polar than Arg, and it is used in Japan for prostate cancer and other applications, such as pancreatitis and liver fibrosis. Consistently, camostat produced a partial block (50–60%) of SARS-CoV-2 entry in TMPRSS2+ cell lines, including Calu-3 lung cells, while no effect was observed in TMPRSS2- cells, and full inhibition was obtained again adding E64d [21].
Interestingly, animal model studies have found that the treatment of camostat mesylate not only produced a 10-fold reduction SARS-CoV titers in Calu-3 airway epithelial cells [69], but also an increase of survival rate of 60% in mice [70]. Camostat can inhibit in vivo infection by SARS-CoV and other pneumoviruses known to utilize TMPRSS2; therefore, the drug could be a suitable antiviral candidate for drug repurposing as component of a drug combination, to prevent infections in the lungs by SARS-CoV-2.
Indeed, camostat has recently been involved in an interventional study to evaluate the efficacy and safety in humans of inhibiting SARS-CoV-2 infection, which provides a randomized treatment in 580 participants with camostat mesylate drug (Phase I) and in parallel with placebo oral tablet (Phase IIa) (ClinicalTrials.gov Identifier: NCT04321096). In order to obtain the highest grade of evidence, double-blinded, randomised, placebo controlled trials are carried out on 334 patients with moderate COVID-19 infection. The clinical trial is in phase IV, but not yet recruiting (ClinicalTrials.gov Identifier: NCT04338906).
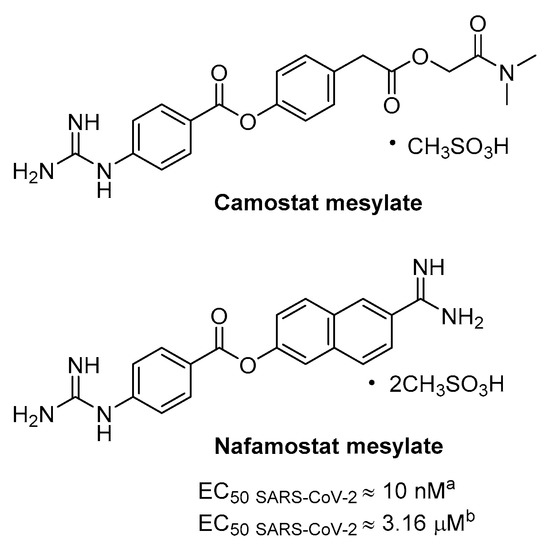
Nafamostat is a serine protease inhibitor that is structurally related to camostat, in use as anticoagulant used for disseminated intravascular coagulation (DIC), that has proven particularly potent activity in blocking CoV infection in vitro likely by inhibiting TMPRSS2 mediated entry (Figure 11). A dual split protein (DSP) reporter assay was developed to quickly monitor membrane fusion mediated by viral S protein and to screen a library of approved drugs, leading to the identification of nafamostat as a potent inhibitor of fusion S activity of MERS. Tested in MERS infection assay the compound blocked viral replication by 100-fold at a very low concentration of 1 nM, more efficiently than camostat [71].
More recently, the same research group has exploited the DSP using SARS-CoV-2 S protein where nafamostat shows fusion inhibitory activity and more interesting the drug inhibits with excellent potency SARS-CoV-2 replication in pulmonary Calu-3 cells with EC50 CPE of about 10 nM with pre-treatment [72]. The activity decreases of more than 300-fold if the inhibitor is added during infection, thus suggesting that it inhibits viral entry. Moreover, nafamostat shows 30–240 nM concentration by iv administration through continuous infusion in DIC patients [72] and PK study in rats revealed the maximum concentration of intact nafamostat in the lung after infusion to be about 60-fold higher in comparison with the maximum blood concentration [73]; such an accumulation may partially suppress SARS-CoV-2 infection.
A randomized clinical trial has recently been launched for adult COVID-19 patients to investigate the ability of nafamostat to slow down the lung disease (ClinicalTrials.gov Identifier: NCT04352400). Indeed, the efficacy of nafamostat as mucolytic and anticoagulant agent and the potent inhibitory activity against TMPRSS2 are useful features to improve the clinical conditions of hospitalized COVID-19 patients.
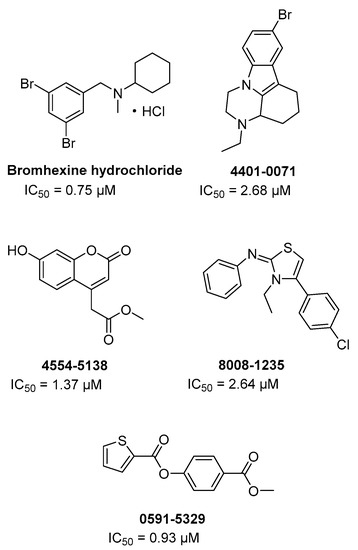
Through a HTS on FDA approved drugs and other commercial libraries (around 70 K cmpds) against TMPRSS2 in order to find new potential anti-metastatic agents for prostate cancer, bromhexine hydrochloride and other four hits (Figure 12) were identified as inhibitors of the enzyme at a concentration below 5 µM [74].
In particular, bromhexine hydrochloride exhibited the most potent inhibition with IC50 = 0.75 µM, resulting specific for TMPRSS2 being significantly less active (50–80-folds) against hepsin and matriptase and not active up to 100 µM against trypsin and thrombin. Moreover, the new repurposed drug was evaluated in cells and in rodent animals without showing significant toxicity.
Bromhexine is structurally unrelated to guanidine derivatives camostat and nafamostat, and no data are provided on kinetics of inhibition and putative binding site. However, bromhexine is an orally bioavailable drug used as mucolytic cough suppressant and with no substantial adverse effects. Indeed, in China an interventional clinical trial for COVID-19 has recently been approved in order to evaluate the efficacy and safety of bromhexine hydrochloride in patients with suspected or novel coronavirus pneumonia (ClinicalTrials.gov Identifier: NCT04273763).
The treatment is randomized open label, based on the administration of bromhexine hydrochloride tablets in combination with standard treatment for COVID-19 (Arbidol hydrochloride granules/Recombinant Human Interferon α2b spray). A larger clinical study on 140 participants, at early phase I, involves the treatment with bromhexine alone or in combination with hydroxychloroquine sulphate, in order to evaluate the effect of bromhexine in preventing the development of COVID-19 (ClinicalTrials.gov Identifier: NCT04340349). In summary, the above-described clinical trials aim to counteract the SARS-CoV-2 infectivity and, in this regard, bromhexine is investigated as a promising inhibitor of TMPRSS2.
A peptidomimetic approach represents a possible alternative towards the development of serine protease TMPRSS2 inhibitors. A series of 4-amidinobenzylamide derivatives, known as inhibitors of various trypsin-like serine protease, was screened against TMPRSS2 [75,76,77] in order to systematically investigate its substrate specificity [78], since the catalytic domain of all trypsin-like serine proteases share structural features and folding pattern.
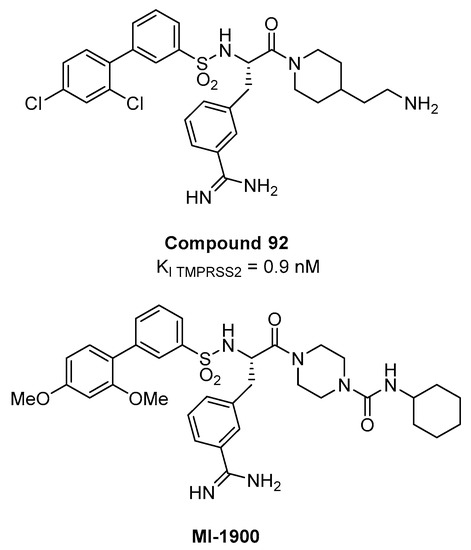
The screening revealed a preference for basic P3 residues in D-configuration, such as D-arginine, proline or glycine residues in P2 position, and a particular preference for 4-amidinophenylalanine amide as P1 residue. Upon SAR investigation, compound 92 (Figure 13), having the P1 m- amidinophenylalanine piperidine amide with a basic ethylamine chain extending towards the S1’ and a bulky biphenyl sulfonamidic N-cap, turned out to be the most potent inhibitor with a Ki value of 0.9 nM for TMPRSS2. In Calu-3 airway epithelial cells that were infected with human pandemic influenza viruses, 92 caused dose-dependent reduction in viral titers (10–100-fold at 10 µM and 100–1000-fold at 50 µM, after 24 h) without affecting cell viability.
However, the significant discrepancies between the nM Ki on the isolated TMPRSS2 and activity in cell-based context observed at µM concentration are likely due to the high polarity of the compound that has two protonable groups at physiological pH. Recently, compound 92 and its less polar analogue MI-1900 have been shown to reduce by 25-fold virus titer in SARS-CoV-2 Calu-3 infected cells at a concentration of 10 µM, without showing toxicity up to 50 µM [79].
In recent times, it has been shown that a guanine-rich tract in the promoter region of human TMPRSS2 gene is able to form G-quadruplex secondary structures in the presence of potassium cations and adjust the gene transcription process [80]. Because the guanine-rich sequence has proven relevant for TMPRSS2 promoter activity [80], the use of compounds that are capable of stabilizing G-quadruplex structures and thus reducing/blocking transcription of the TMPRSS2 gene has been proposed as a potential host targeting strategy.
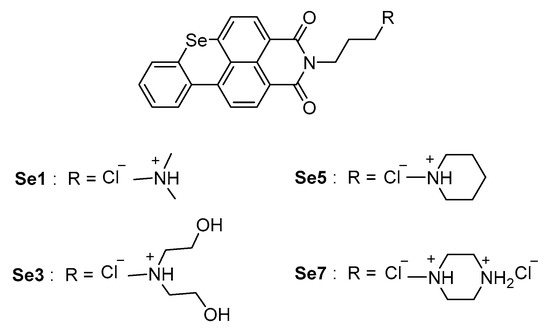
Seven benzoselenoxanthene analogues were designed and synthesized for this purpose, and the compounds Se1, Se3, Se5, and Se7 (Figure 14) have shown to increase the stability of the TMPRSS2 G-quadruplex in vitro, corresponding to an effective decrease of TMPRSS2-gene expression in Calu-3 cells. At a later stage, Shen et al. evaluated the inhibition of viral propagation in Calu-3 cells infected with Influenza A virus.
Benzoselenoxanthene analogues led to a near-complete reduction of virus titer at a concentration of 8 µM, with an antiviral activity comparable with anti-influenza drug Oseltamivir, although definitely inferior to camostat inhibitor. Therefore, no significant cytotoxic effects have been revealed at 10 µM. In summary, the down-regulation of TMPRSS2 expression through G-quadruplex structure stabilization is affirmed as a promising strategy for the inhibition of viral infection and represents a pioneering starting point for novel drugs against SARS-CoV-2.
reference link : https://www.mdpi.com/1422-0067/21/16/5707/htm
More information: Rina Hashimoto et al, Dual inhibition of TMPRSS2 and Cathepsin B prevents SARS-CoV-2 infection in iPS cells, Molecular Therapy – Nucleic Acids (2021). DOI: doi.org/10.1016/j.omtn.2021.10.016



{kind=link}