











 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Il vaccino COVID-19 è solo un esempio dello sforzo rapido e globale per fermare la pandemia. Anche i farmaci sono in fase di sviluppo. Un nuovo studio dei ricercatori del CiRA mostra che la combinazione di due farmaci blocca l’infezione di SARS-CoV-2, il virus responsabile del COVID-19, nelle cellule iPS.
L’influenza della pandemia di COVID-19 può essere vista in quasi tutti i settori. Le forniture a catena stanno soffrendo in tutto il mondo e le aziende di tutto il mondo si stanno abituando a far lavorare i propri dipendenti da casa. Anche la scienza è stata influenzata in molti modi.
CiRA nel suo insieme ha deviato enormi risorse al problema, perché le cellule iPS offrono un modello interessante per studiare l’infezione per diversi motivi.
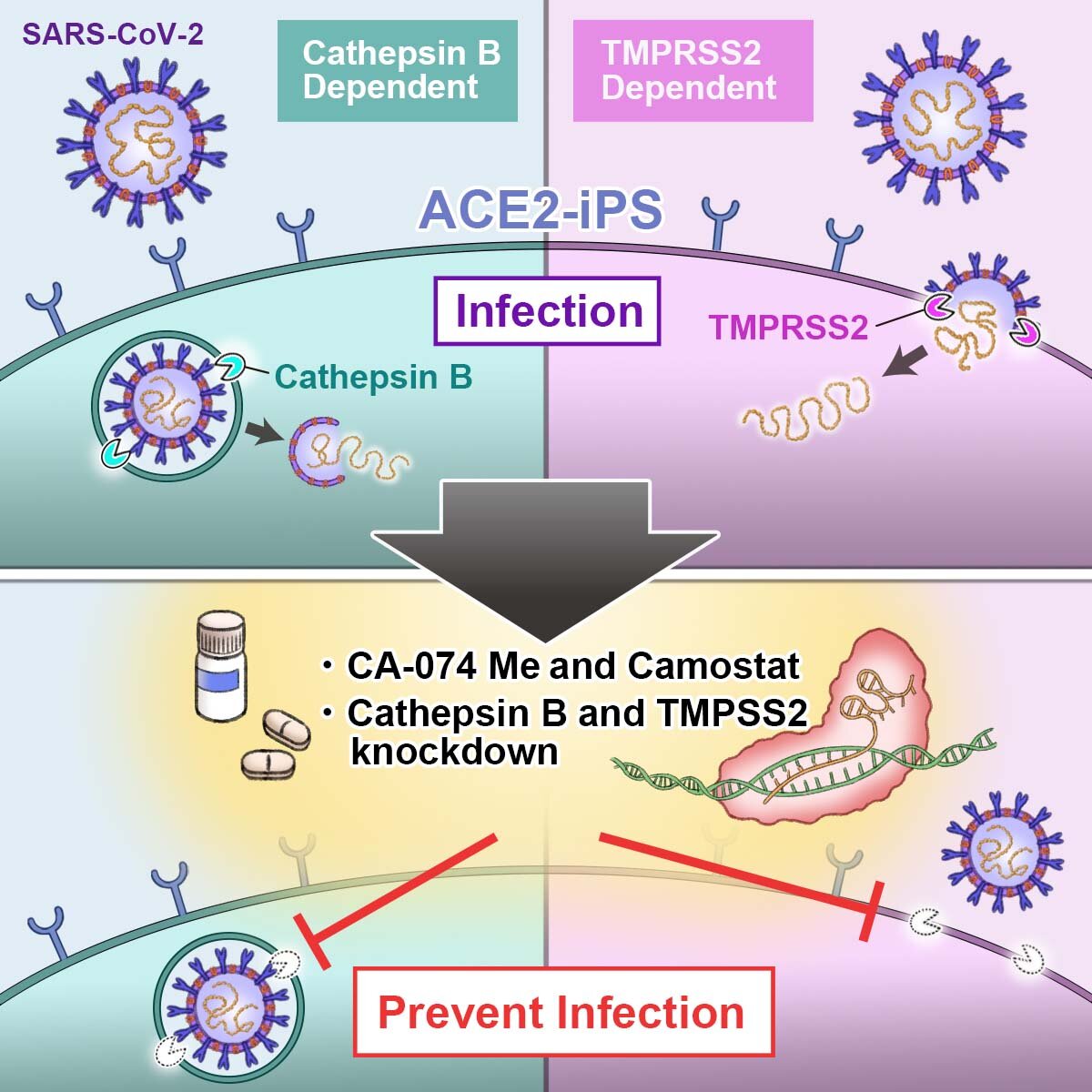
“Possiamo differenziare le cellule iPS in qualsiasi tipo di cellula che vogliamo studiare. Possiamo acquisire cellule iPS da pazienti COVID-19 lievi e gravi”, ha affermato Kazuo Takayama, professore associato junior del CiRA, su alcuni dei vantaggi di queste cellule.
Nello studio, questi due geni sono stati modificati in modo che la cellula non producesse le proteasi. Ciò ha significativamente attenuato l’infezione delle cellule iPS da SARS-CoV-2.
Utilizzando queste informazioni, i ricercatori hanno testato due farmaci, CA-074, che inibisce la catepsina B, e Camostat, che inibisce la serina proteasi 2 transmembrana, dimostrando che la loro combinazione ha ridotto la carica virale a meno dello 0,01% di quella senza trattamento farmacologico.
Questa sinergia potrebbe riflettere le diverse posizioni delle proteasi nella cellula. Cathespin B si trova negli endosomi, mentre TMPRSS2 si trova nella membrana cellulare. Gli endosomi sono molecole da trasportare da una posizione all’altra in una cellula, mentre le proteine nella membrana possono muoversi ma solo sulla membrana e scambiare oggetti dall’interno e dall’esterno della cellula.
Tuttavia, Takayama avverte che le cellule iPS non esistono nei pazienti e che sono necessari ulteriori studi per confermare l’effetto della combinazione di farmaci nella cura del paziente.
“Dobbiamo vedere se gli stessi effetti si trovano nelle cellule iPS differenziate”, ha detto.
Targeting della proteasi dell’host per bloccare l’ingresso virale
La scissione proteolitica della proteina S nei siti S1/S2 e S2′ da parte della serina proteasi TMPRRS2 e/o della cisteina proteasi endosomiale CatB/L guida l’ingresso del virus attraverso il peptide di fusione che si inserisce nella membrana della cellula ospite. Questo inserimento porta alla formazione di un fascio a sei eliche antiparallelo, consentendo il processo di fusione e, quindi, lo svestimento e il rilascio dell’RNA virale nel citoplasma.
Gli eventi di priming S1/S2 e S2′ da parte delle proteasi dell’ospite sono necessari affinché SARS-CoV-2 infetti l’ospite e l’interferenza con l’ingresso del virus potrebbe rivelarsi una strategia antivirale vantaggiosa, poiché bloccherebbe l’infezione o la propagazione del virus a una fase iniziale, cosa ancora più importante se si considera la sua elevata trasmissibilità. Mirare ai fattori dell’ospite ha il vantaggio di ridurre il possibile sviluppo di resistenza ai farmaci e di fornire probabilmente un’attività ad ampio spettro, al contrario interagendo con la proteina dell’ospite, la possibilità di avere effetti collaterali più gravi è maggiore rispetto all’approccio antivirale classico.
Tuttavia, il coinvolgimento di TMPRRS2 e CatB/L nell’infezione virale è ancora oggetto di studio e il targeting di queste proteasi dell’ospite per il trattamento del CoV è una strategia emergente. Inoltre, i dati disponibili suggeriscono che è necessaria l’inibizione simultanea di entrambe le proteasi per un solido blocco dell’ingresso antivirale [58,59].
Finora, non ci sono segnalazioni di programmi di chimica farmaceutica incentrati su TMPRRS2 e catepsina B/L all’interno della scoperta di farmaci CoV, quindi gli inibitori disponibili spesso mostrano limitazioni, in termini di potenza, selettività, proprietà simili a farmaci. Tuttavia, è stato dimostrato che alcuni composti esercitano un promettente effetto antivirale contro SARS-CoV-2 e/o altri CoV correlati e sono descritti di seguito.
TMPRSS2 come target host e suoi inibitori
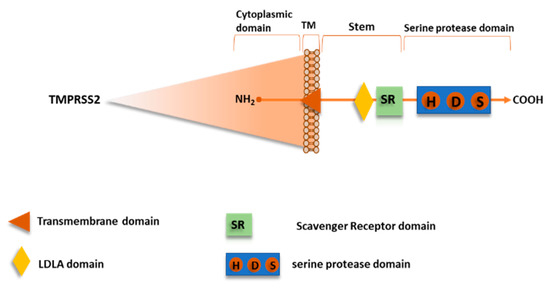
Recentemente, è stato dimostrato che TMPRSS2 media il priming della proteina SARS-CoV-2 S, nonché per SARS-CoV e altri CoV [21,58,60,61,62]. TMPRSS2, chiamato anche Epitheliasin, è una serina proteasi 492 aa della famiglia delle serina proteasi transmembrana di tipo II, espresse sulla superficie cellulare, coerentemente al loro ruolo nella regolazione delle interazioni cellula-cellula e cellula-matrice. La famiglia umana TTSP comprende 17 membri che finora condividono le stesse caratteristiche strutturali.
Il dominio intracellulare N-terminale conserva i siti di fosforilazione, seguito dal dominio transmembrana e la regione dello stelo che si trova nella parte extracellulare iniziale con un sito di legame per lipoproteine a bassa densità (LDL) e calcio in un motivo del recettore A LDL e un singolo recettore scavenger ricco di Cys (dominio SRCR). Il dominio endoproteasico extracellulare C-terminale contiene la triade catalitica composta dai residui His-Asp-Ser, in cui il gruppo ossidrile Ser promuove l’attacco nucleofilo al sito di priming (Figura 10) [63].
TMPRSS2 è espresso prevalentemente nella prostata, ma è stato trovato anche nei polmoni, nel colon, nel fegato, nei reni e nel pancreas. L’espressione nelle vie aeree superiori, nei bronchi e nei polmoni, dove la sua funzione fisiologica rimane poco chiara, fa pensare al suo ruolo importante per il pneumotropismo di diversi virus altamente patogeni, come SARS-CoV-2, SARS-CoV, MERS-CoV e HCoV -NL63.
Infatti, i CoV impegnano ACE2 per entrare nelle cellule ospiti e sebbene l’ACE2 sia un enzima ubiquitario, mostrano un particolare tropismo per i polmoni. Inoltre, mentre ACE2 è espresso sia nei pneumociti di tipo I che di tipo II, è stato verificato che SARS-CoV infetta prontamente i pneumociti di tipo I in fase iniziale [60,64]. Esperimenti in vivo hanno mostrato che TMPRSS2 è responsabile della diffusione virale e dell’immunopatologia dell’infezione da CoV [59].
Nei topi TMPRSS2_ko, SARS-CoV e MERS-CoV hanno mostrato una replicazione virale significativamente ridotta nei polmoni, specialmente nei bronchioli, e l’infiltrazione infiammatoria. Inoltre, TMPRSS2 non è solo coinvolto nell’attivazione della proteina CoVs S, ma il suo ruolo è stato anche riconosciuto nell’attivazione della glicoproteina sulla superficie del virus dell’influenza A, del metapneumovirus e del virus della diarrea epidemica suina in diverse fasi dei cicli di vita virali [65] .
Pertanto, prendere di mira TMPRSS2 potrebbe essere una strategia antivirale ad ampio spettro; tuttavia, non sono stati identificati farmaci in grado di inibire specificamente questo bersaglio, né sono disponibili informazioni sufficienti sulla specificità del substrato e non sono disponibili strutture 3D della proteina. Tuttavia, è stato riportato che i substrati fluorogenici della tripsina Cbz-Gly-Gly-Arg-AMC [66] e Boc-Leu-Gly-Arg-AMC [67] erano anche substrati per TMPRSS2, indicando che P1 può essere rappresentato da Arg, che è coerente con gli elementi di riconoscimento di alcuni farmaci in grado di inibire le serina proteasi dell’epitelio umano; tuttavia non sono state effettuate analisi di cinetica enzimatica e non sono noti i valori di Km o Kcat [67]. D’altra parte, farmaci in grado di inibire un ampio pannello di serina proteasi umane, tra cui TMPRSS2,
Precedenti evidenze hanno mostrato che l’inibitore della serina proteasi camostat mesilato è stato in grado, anche ad alte concentrazioni (fino a 100 µM), di bloccare parzialmente l’infezione da SARS-CoV (65% di inibizione) nelle cellule che esprimono TMPRSS2 senza causare tossicità; quindi, l’attività antivirale è stata potenziata con l’aggiunta di un inibitore CatB/L, vale a dire E64d, indicando così che il restante 35% era attribuibile alle catepsine endosomiali [58].
Camostat è un inibitore pseudo irreversibile di diverse serina proteasi, tra cui TMPRSS2, essendo caratterizzato da guanidina aromatica come elementi di riconoscimento mimetici P1 (Figura 11), meno polare di Arg, ed è utilizzato in Giappone per il cancro alla prostata e altre applicazioni, come la pancreatite e fibrosi epatica. Coerentemente, camostat ha prodotto un blocco parziale (50-60%) dell’ingresso di SARS-CoV-2 nelle linee cellulari TMPRSS2+, comprese le cellule polmonari Calu-3, mentre non è stato osservato alcun effetto nelle cellule TMPRSS2- e l’inibizione completa è stata ottenuta nuovamente aggiungendo E64d [21].
È interessante notare che studi su modelli animali hanno scoperto che il trattamento del mesilato camostat non solo ha prodotto una riduzione di 10 volte dei titoli SARS-CoV nelle cellule epiteliali delle vie aeree Calu-3 [69], ma anche un aumento del tasso di sopravvivenza del 60% nei topi [70 ]. Camostat può inibire l’infezione in vivo da SARS-CoV e altri pneumovirus noti per utilizzare TMPRSS2; pertanto, il farmaco potrebbe essere un candidato antivirale adatto per il riutilizzo di farmaci come componente di una combinazione di farmaci, per prevenire le infezioni polmonari da SARS-CoV-2.
Infatti, camostat è stato recentemente coinvolto in uno studio interventistico per valutare l’efficacia e la sicurezza nell’uomo dell’inibizione dell’infezione da SARS-CoV-2, che prevede un trattamento randomizzato in 580 partecipanti con farmaco mesilato camostat (Fase I) e in parallelo con placebo orale tablet (Fase IIa) (ClinicalTrials.gov Identifier: NCT04321096). Al fine di ottenere il più alto grado di evidenza, vengono condotti studi in doppio cieco, randomizzati, controllati con placebo su 334 pazienti con infezione da COVID-19 moderata. La sperimentazione clinica è in fase IV, ma non è ancora in fase di reclutamento (ClinicalTrials.gov Identifier: NCT04338906).
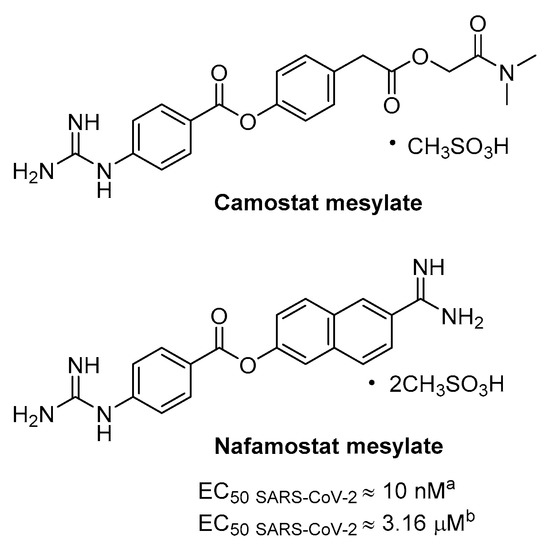
Nafamostat è un inibitore della serina proteasi strutturalmente correlato al camostat, in uso come anticoagulante utilizzato per la coagulazione intravascolare disseminata (DIC), che ha dimostrato un’attività particolarmente potente nel bloccare l’infezione da CoV in vitro probabilmente inibendo l’ingresso mediato da TMPRSS2 (Figura 11). È stato sviluppato un test reporter a doppia proteina split (DSP) per monitorare rapidamente la fusione della membrana mediata dalla proteina S virale e per lo screening di una libreria di farmaci approvati, portando all’identificazione di nafamostat come potente inibitore dell’attività di fusione S di MERS. Testato nel test di infezione da MERS, il composto ha bloccato la replicazione virale di 100 volte a una concentrazione molto bassa di 1 nM, in modo più efficiente del camostat [71].
Più recentemente, lo stesso gruppo di ricerca ha sfruttato il DSP utilizzando la proteina SARS-CoV-2 S dove nafamostat mostra attività inibitoria della fusione e più interessante il farmaco inibisce con eccellente potenza la replicazione di SARS-CoV-2 nelle cellule polmonari Calu-3 con EC50 CPE di circa 10 nM con pretrattamento [72]. L’attività diminuisce di oltre 300 volte se l’inibitore viene aggiunto durante l’infezione, suggerendo così che inibisce l’ingresso del virus. Inoltre, nafamostat mostra una concentrazione di 30-240 nM per somministrazione ev attraverso infusione continua nei pazienti con DIC [72] e lo studio PK nei ratti ha rivelato che la concentrazione massima di nafamostat intatto nel polmone dopo l’infusione è circa 60 volte più alta rispetto al massimo concentrazione ematica [73]; tale accumulo può sopprimere parzialmente l’infezione da SARS-CoV-2.
Recentemente è stato avviato uno studio clinico randomizzato per pazienti adulti COVID-19 per studiare la capacità del nafamostat di rallentare la malattia polmonare (identificatore ClinicalTrials.gov: NCT04352400). Infatti, l’efficacia del nafamostat come agente mucolitico e anticoagulante e la potente attività inibitoria contro TMPRSS2 sono caratteristiche utili per migliorare le condizioni cliniche dei pazienti COVID-19 ospedalizzati.
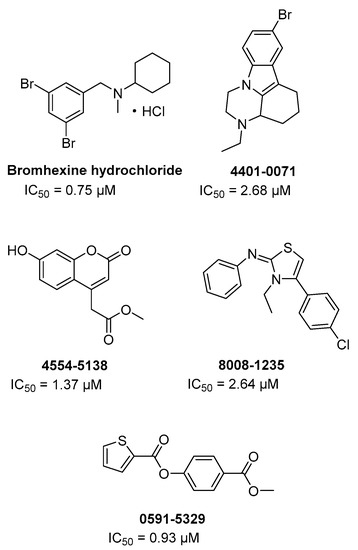
Attraverso un HTS su farmaci approvati dalla FDA e altre librerie commerciali (circa 70 K cmpds) contro TMPRSS2 al fine di trovare nuovi potenziali agenti antimetastatici per il cancro alla prostata, la bromexina cloridrato e altri quattro hit (Figura 12) sono stati identificati come inibitori dell’enzima ad una concentrazione inferiore a 5 µM [74].
In particolare, la bromexina cloridrato ha mostrato l’inibizione più potente con IC50 = 0,75 µM, risultando specifica per TMPRSS2 essendo significativamente meno attiva (50-80 volte) contro epsina e matriptasi e non attiva fino a 100 µM contro tripsina e trombina. Inoltre, il nuovo farmaco riproposto è stato valutato nelle cellule e negli animali roditori senza mostrare una tossicità significativa.
La bromexina non è strutturalmente correlata ai derivati della guanidina camostat e nafamostat e non sono forniti dati sulla cinetica dell’inibizione e sul sito di legame putativo. Tuttavia, la bromexina è un farmaco biodisponibile per via orale utilizzato come soppressore della tosse mucolitico e senza effetti collaterali sostanziali. Infatti, in Cina è stato recentemente approvato uno studio clinico interventistico per COVID-19 al fine di valutare l’efficacia e la sicurezza della bromexina cloridrato in pazienti con polmonite da coronavirus sospetta o nuova (ClinicalTrials.gov Identifier: NCT04273763).
Il trattamento è randomizzato in aperto, basato sulla somministrazione di compresse di bromexina cloridrato in combinazione con il trattamento standard per COVID-19 (granuli di cloridrato di Arbidol/interferone umano ricombinante α2b spray). Uno studio clinico più ampio su 140 partecipanti, nella fase iniziale I, prevede il trattamento con bromexina da sola o in combinazione con idrossiclorochina solfato, al fine di valutare l’effetto della bromexina nella prevenzione dello sviluppo di COVID-19 (Identificatore ClinicalTrials.gov: NCT04340349) . In sintesi, gli studi clinici sopra descritti mirano a contrastare l’infettività SARS-CoV-2 e, a questo proposito, la bromexina è studiata come un promettente inibitore di TMPRSS2.
Un approccio peptidomimetico rappresenta una possibile alternativa allo sviluppo di inibitori della serina proteasi TMPRSS2. Una serie di derivati 4-amidinobenzilammide, noti come inibitori di varie serina proteasi simili alla tripsina, è stata sottoposta a screening contro TMPRSS2 [75,76,77] al fine di indagare sistematicamente la sua specificità di substrato [78], poiché il dominio catalitico di tutte le tripsina- come le serina proteasi condividono le caratteristiche strutturali e il modello di piegatura.
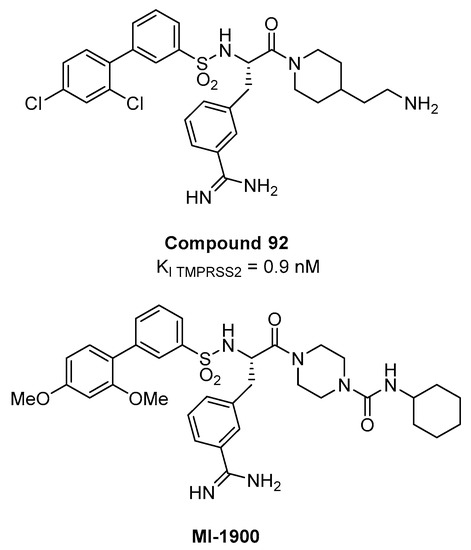
Lo screening ha rivelato una preferenza per i residui di base P3 in configurazione D, come residui di D-arginina, prolina o glicina in posizione P2, e una particolare preferenza per l’ammide 4-amidinofenilalanina come residuo P1. Dall’indagine SAR, il composto 92 (Figura 13), avente la P1 m-ammidinofenilalanina piperidina ammide con una catena etilammina basica che si estende verso S1′ e un voluminoso N-cap bifenil sulfonamidico, è risultato essere l’inibitore più potente con un valore di Ki di 0,9 nM per TMPRSS2. Nelle cellule epiteliali delle vie aeree Calu-3 che sono state infettate da virus dell’influenza pandemica umana, 92 hanno causato una riduzione dose-dipendente dei titoli virali (10-100 volte a 10 µM e 100-1000 volte a 50 µM, dopo 24 h) senza influenzare vitalità cellulare.
Tuttavia, le discrepanze significative tra il nM Ki sul TMPRSS2 isolato e l’attività nel contesto cellulare osservato alla concentrazione µM sono probabilmente dovute all’elevata polarità del composto che ha due gruppi protonabili a pH fisiologico. Recentemente, il composto 92 e il suo analogo meno polare MI-1900 hanno dimostrato di ridurre di 25 volte il titolo del virus nelle cellule infette da SARS-CoV-2 Calu-3 a una concentrazione di 10 µM, senza mostrare tossicità fino a 50 µM [79 ].
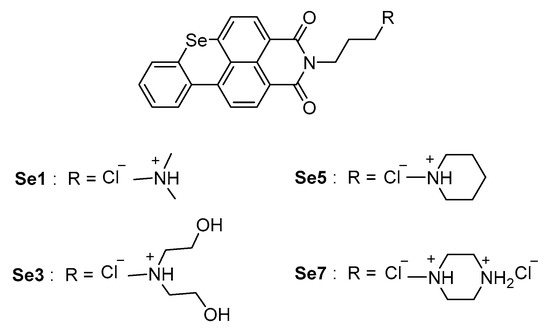
In tempi recenti, è stato dimostrato che un tratto ricco di guanina nella regione del promotore del gene TMPRSS2 umano è in grado di formare strutture secondarie G-quadruplex in presenza di cationi di potassio e regolare il processo di trascrizione genica [80]. Poiché la sequenza ricca di guanina si è dimostrata rilevante per l’attività del promotore TMPRSS2 [80], l’uso di composti in grado di stabilizzare le strutture G-quadruplex e quindi di ridurre/bloccare la trascrizione del gene TMPRSS2 è stato proposto come potenziale strategia di targeting dell’ospite.
A questo scopo sono stati progettati e sintetizzati sette analoghi del benzoselenoxantene e i composti Se1, Se3, Se5 e Se7 (Figura 14) hanno dimostrato di aumentare la stabilità del G-quadruplex TMPRSS2 in vitro, corrispondente a un’effettiva diminuzione del gene TMPRSS2 espressione nelle cellule Calu-3. In una fase successiva, Shen et al. valutato l’inibizione della propagazione virale nelle cellule Calu-3 infettate dal virus dell’influenza A.
Gli analoghi del benzoselenoxantene hanno portato ad una riduzione quasi completa del titolo virale alla concentrazione di 8 µM, con un’attività antivirale paragonabile al farmaco antinfluenzale Oseltamivir, sebbene decisamente inferiore all’inibitore del camostat. Pertanto, non sono stati rilevati effetti citotossici significativi a 10 µM. In sintesi, la down-regulation dell’espressione di TMPRSS2 attraverso la stabilizzazione della struttura G-quadruplex si afferma come una strategia promettente per l’inibizione dell’infezione virale e rappresenta un punto di partenza pionieristico per nuovi farmaci contro SARS-CoV-2.
collegamento di riferimento: https://www.mdpi.com/1422-0067/21/16/5707/htm
Maggiori informazioni: Rina Hashimoto et al, La doppia inibizione di TMPRSS2 e Catepsina B previene l’infezione da SARS-CoV-2 nelle cellule iPS, Terapia molecolare – Acidi nucleici (2021). DOI: doi.org/10.1016/j.omtn.2021.10.016



{kind=link}