










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



B.1.620 Lineage: According to a new multinational research led by researchers from Lithuania, a new SARS-CoV-2 Lineage called B.1.620 has emerged from most probably Cameroon in Central Africa and has spreading silently across the globe silently while the world was more focused on the Delta variant and its emerging sub-variants.
This new immune evasive variant B.1.620 not only exhibits the spike protein mutation E484K but also carries a large number of unique mutations.
In fact, this new SARS-CoV-2 strain is 18 mutations away from nearest relatives and 26 from the reference strain Wuhan-Hu-1.
This worrisome B.1.620 variant shares multiple mutations and deletions with known SARS-CoV-2 variants of concern (VOCs) including HV69/70Δ, LLA241/243Δ, S477N, E484K, and P681H. However, it does not appear to be of recombinant origin.
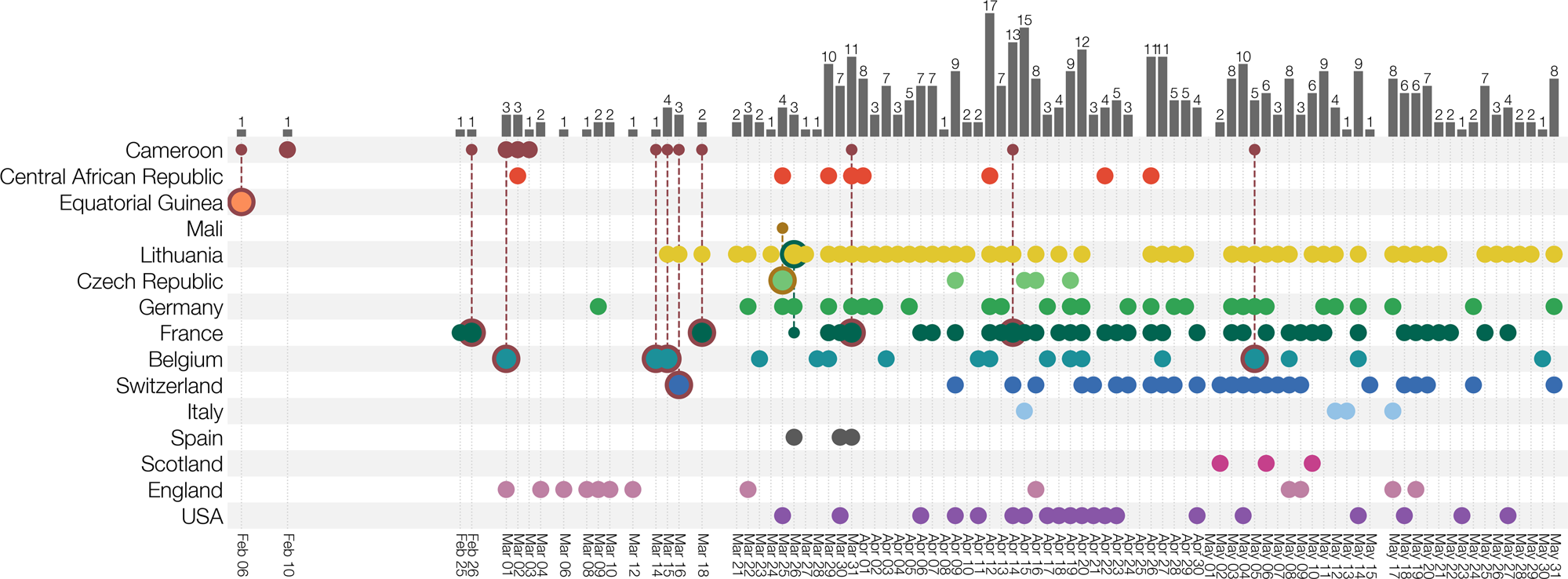
This new lineage now includes genomes from France, Switzerland, Belgium, Germany, England, Scotland, Italy, Spain, Czechia, Norway, Sweden, Ireland, Portugal, the United States, Canada, and, most recently, the Philippines and South Korea. Initial B.1.620 European cases included travelers returning from Cameroon; however, more recently, genomes are also being submitted to GISAID from the Central African Republic, Equatorial Guinea, the Democratic Republic of the Congo, Gabon, and the Republic of Congo.
It is currently causing a COVID-19 resurgence in Lithuania with worrying more critical outcomes.
In the study abstract, the researchers commented, “Distinct SARS-CoV-2 lineages, discovered through various genomic surveillance initiatives, have emerged during the pandemic following unprecedented reductions in worldwide human mobility. We here describe a SARS-CoV-2 lineage – designated B.1.620, discovered in Lithuania and carrying many mutations and deletions in the spike protein shared with widespread variants of concern (VOCs), including E484K, S477N and deletions HV69Δ, Y144Δ, and LLA241/243Δ.
As well as documenting the suite of mutations this lineage carries, we also describe its potential to be resistant to neutralizing antibodies, accompanying travel histories for a subset of European cases, evidence of local B.1.620 transmission in Europe with a focus on Lithuania, and significance of its prevalence in Central Africa owing to recent genome sequencing efforts there. We make a case for its likely Central African origin using advanced phylogeographic inference methodologies incorporating recorded travel histories of infected travellers.”
The study findings were published in the peer reviewed journal: Nature.
https://www.nature.com/articles/s41467-021-26055-8
The WHO and U.S.CDC has been notified of the recent developments and the B.1.620 is likely to be upgraded to a VOC (Variant of concern) status in coming days.
Over a year into the pandemic and with an unprecedented reduction in human mobility worldwide, distinct SARS-CoV-2 lineages have arisen in multiple geographic areas around the world1,2,3. New lineages are constantly appearing (and disappearing) all over the world and may be designated variant under investigation (VUI) if considered to have concerning epidemiological, immunological or pathogenic properties.
So far, four lineages (i.e. B.1.1.7, B.1.351, P.1 and B.1.617.2 according to the Pango SARS-CoV-2 lineage nomenclature4,5) have been universally categorised as variants of concern (VOCs), due to evidence of increased transmissibility, disease severity and/or possible reduced vaccine efficacy. An even broader category termed variant of interest (VOI) encompasses lineages that are suspected to have an altered phenotype implied by their mutation profile.
In some cases, a lineage may rise to high frequency in one location and seed others in its vicinity, such as lineage B.1.177 that became prevalent in Spain and was later spread across the rest of Europe2. In others, reductions in human mobility, insufficient surveillance and passage of time allowed lineages to emerge and rise to high frequency in certain areas, as has happened with lineage A.23.1 in Uganda6, a pattern reminiscent of holdover H1N1 lineages discovered in West Africa years after the 2009 pandemic7.
In the absence of routine genomic surveillance at their origin location, diverged lineages may still be observed as travel cases or transmission chains sparked by such in countries that do have sequencing programmes in place. A unique SARS-CoV-2 variant found in Iran early in the pandemic was characterised in this way8, and recently travellers returning from Tanzania were found to be infected with a lineage bearing multiple amino acid changes of concern9.
As more countries launch their own SARS-CoV-2 sequencing programmes, introduced strains are easier to detect since they tend to be atypical of a host country’s endemic SARS-CoV-2 diversity, particularly so when introduced lineages have accumulated genetic diversity not observed previously, a phenomenon that is characterised by long branches in phylogenetic trees.
In Rwanda, this was exemplified by the detection of lineage B.1.3806, which was characteristic of Rwandan and Ugandan epidemics at the time. The same sequencing programme was then perfectly positioned to observe a sweep where B.1.380 was replaced by lineage A.23.16, which was first detected in Uganda10, and to detect the country’s first cases of B.1.1.7 and B.1.351. Similarly, sequencing programmes in Europe were witness to the rapid displacement of pan-European and endemic lineages with VOCs, primarily B.1.1.7 (e.g. Lyngse et al.11).
Given the appearance of VOCs towards the end of 2020 and the continued detection of previously unobserved SARS-CoV-2 diversity, it stands to reason that more variants of interest (VOIs), and perhaps even VOCs, can and likely do circulate in areas of the world where access to genome sequencing is not available nor provided as a service by international organisations.
Lineage A.23.110 from Uganda and a provisionally designated variant of interest A.VOI.V29 from Tanzania might represent the first detections of a much more diverse pool of variants circulating in Africa. We here describe a similar case in the form of a lineage designated B.1.620 that first caught our attention as a result of what was initially a small outbreak caused by a distinct and diverged lineage previously not detected in Lithuania, bearing multiple VOC-like mutations and deletions, many of which substantially alter the spike protein.
The first samples of B.1.620 in Lithuania were redirected to sequencing because they were flagged by occasional targeted PCR testing for SARS-CoV-2 spike protein mutation E484K repeated on PCR-positive samples. Starting April 2nd 2021, targeted E484K PCR confirmed a growing cluster of cases with this mutation in Anykščiai municipality in Utena county with a total of 43 E484K+ cases out of 81 tested by April 28th (Supplementary Fig. S1).
Up to this point, the Lithuanian genomic surveillance programme had sequenced over 10% of PCR-positive SARS-CoV-2 cases in Lithuania and identified few lineages with E484K circulating in Lithuania. During initial B.1.620 circulation in Lithuania the only other E484K-bearing lineages in Lithuania had been B.1.351 (one isolated case in Kaunas county, and 12 cases from a transmission chain centred in Vilnius county) and B.1.1.318 (one isolated case in Alytus county), none of which had been found in Utena county despite a high epidemic sequencing coverage in Lithuania (Supplementary Fig. S2).
An in-depth search for relatives of this lineage on GISAID12 uncovered a few genomes from Europe initially, though more continue to be found since B.1.620 received its Pango lineage designation which was subsequently integrated into GISAID. This lineage now includes genomes from a number of European countries such as France, Switzerland, Belgium, Germany, England, Scotland, Italy, Spain, Czechia, Norway, Sweden, Ireland, and Portugal, North America: the United States (US) and Canada, and most recently The Philippines and South Korea in Asia.
Interestingly, a considerable proportion of initial European cases turned out to be travellers returning from Cameroon. Since late April 2021, sequencing teams operating in central Africa, primarily working on samples from the Central African Republic, Equatorial Guinea, the Democratic Republic of the Congo, Gabon and lately the Republic of Congo have been submitting B.1.620 genomes to GISAID.
We here describe the mutations and deletions the B.1.620 lineage carries, many of which were previously observed in individual VOCs, but not in combination, and present evidence that this lineage likely originated in central Africa and is likely to circulate in the wider region where its prevalence is expected to be high.
By combining collected travel records from infected patients entering different European countries, and by exploiting this information in a recently developed Bayesian phylogeographic inference methodology13,14, we reconstruct the dispersal of lineage B.1.620 from its inferred origin in the Central African Republic to several of its neighbouring countries, Europe and the US. Finally, we provide a description of local transmission in Lithuania, France, Spain, Italy, and Germany through phylogenetic and phylogeographic analysis, and in Belgium through the collection of travel records.



{kind=link}