










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Israeli researchers from Tel Aviv University have successfully identified 3 key proteins out of the total of 29 proteins found in the SAR-CoV-2 genome that can damage the blood vessels of the human host. These three key proteins were nsp2, nsp5_c145a (catalytic dead mutant of nsp5), and nsp7.
To date it is known that the SARS-CoV-2 genome encodes 29 proteins, whose contribution to the disease manifestations, and especially endothelial complications, is unknown.
The study team cloned and expressed 26 of these proteins in human cells and characterized the endothelial response to overexpression of each, individually.
The researchers showed that whereas most SARS-CoV-2 proteins induced significant changes in endothelial permeability, nsp2, nsp5_c145a (catalytic dead mutant of nsp5), and nsp7 also reduced CD31, and increased von Willebrand factor expression and IL-6, suggesting endothelial dysfunction.
Utilizing propagation-based analysis of a protein–protein interaction (PPI) network, the study team predicted the endothelial proteins affected by the viral proteins that potentially mediate these effects.
The team further applied the PPI model to identify the role of each SARS-CoV-2 protein in other tissues affected by coronavirus disease (COVID-19).
While validating the PPI network model, the study team found that the tight junction (TJ) proteins cadherin-5, ZO-1, and β-catenin are affected by nsp2, nsp5_c145a, and nsp7 consistent with the model prediction.
The study findings identify the SARS-CoV-2 proteins that might be most detrimental in terms of endothelial dysfunction, thereby shedding light on vascular aspects of COVID-19.
The study findings were published in the peer reviewed journal: eLIfe
https://elifesciences.org/articles/69314
Coronavirus disease (COVID-19) caused by the 2019 novel coronavirus (2019-nCoV/SARS-CoV-2) led to a global pandemic in 2020. By late September 2021, coronavirus had infected more than 220 million people worldwide, causing over 4.5 million deaths.
After the initial phase of the viral infection, ~ 30% of patients hospitalized with COVID-19 develop severe disease with progressive lung damage, known as severe acute respiratory syndrome (SARS), and a severe immune response. Interestingly, additional pathologies have been observed, such as hypoxemia and cytokine storm which, in some cases, lead to heart and kidney failure, and neurological symptoms.
Recent observations suggest that these pathologies are mainly due to increased coagulation and vascular dysfunction (Lee et al., 2021; Libby and Lüscher, 2020; Siddiqi et al., 2020). It is currently believed that in addition to being a respiratory disease, COVID-19 might also be a ‘vascular disease’ (Lee et al., 2021), as it may result in a leaky vascular barrier and increased expression of von Willebrand factor (VWF) (Siddiqi et al., 2020), responsible for increased coagulation, cytokine release, and inflammation (Siddiqi et al., 2020; Teuwen et al., 2020; Aid et al., 2020; Potus et al., 2020; Wazny et al., 2020; Pum et al., 2021; Barbosa et al., 2021; Lin et al., 2020; Matarese et al., 2020; Xiao et al., 2020).
Recent studies suggest that the main mechanism disrupting the endothelial barrier occurs in several stages: First, a direct effect on the endothelial cells that causes an immune response of the vascular endothelium (endotheliitis) and endothelial dysfunction.
Second, lysis and death of the endothelial cells Teuwen et al., 2020; Xiao et al., 2020 followed by sequestering of human angiotensin I-converting enzyme 2 (hACE2) by viral spike proteins that activate the kallikrein–bradykinin and renin–angiotensin pathways, increasing vascular permeability (Teuwen et al., 2020; Varga et al., 2020).
Last, overreaction of the immune system, during which a combination of neutrophils and immune cells producing reactive oxygen species, inflammatory cytokines (e.g., interleukin [IL]-1β, IL-6, and tumor necrosis factor), and vasoactive molecules (e.g., thrombin, histamine, thromboxane A2, and vascular endothelial growth factor), and the deposition of hyaluronic acid lead to disruption of endothelial junctions, increased vascular permeability, and leakage and coagulation (Libby and Lüscher, 2020; Teuwen et al., 2020; Varga et al., 2020).
Of great interest is the effect on the brain’s vascular system. Cerebrovascular effects have been suggested to be among the long-lasting effects of COVID-19. Indeed, the susceptibility of brain endothelial cells to direct SARS-CoV-2 infection was found to increase due to increased expression of hACE2 in a blood flow-dependent manner, leading to a unique gene expression process that might contribute to the cerebrovascular effects of the virus (Pober and Sessa, 2007).
While many studies point out the importance of the vascular system in COVID-19 (Kaneko et al., 2021; Jung et al., 2020b; Nägele et al., 2020), only a few Pons et al., 2020; Chioh et al., 2020; Nascimento Conde et al., 2020; Buzhdygan et al., 2020 have looked at the direct vascular response to the virus.
Most of those reports stem from either clinical observations, or in vitro studies or in vivo studies in which animals/cells were transfected with the SARS-CoV-2 virus and their systemic cellular response assessed, without pinpointing the specific viral protein(s) causing the observed changes. SARS-CoV-2 is an enveloped virus with a positive-sense, single-stranded RNA genome of ∼30 kb, encoding 29 proteins (Figure 1).
These proteins can be classified as: structural proteins: S (spike proteins), E (envelope proteins), M (membrane proteins), N (nucleocapsid protein and viral RNA); nonstructural proteins: nsp1–16; open reading frame accessory proteins: orf3–10 (Kim et al., 2020; Hu et al., 2021). Table 1 summarizes the known effects of specific SARS-CoV-2 proteins (Gordon et al., 2020; Peng et al., 2020b; Procko, 2020; Cornillez-Ty et al., 2009; Romano et al., 2020; Hillen et al., 2020; Chi et al., 2003).
The functionality of some of these is still unknown. Moreover, a considerable knowledge gap still exists regarding molecular mechanisms, especially the protein–protein interaction (PPI) pathways (Cowen et al., 2017), leading to tissue dysfunction.
Figure 1
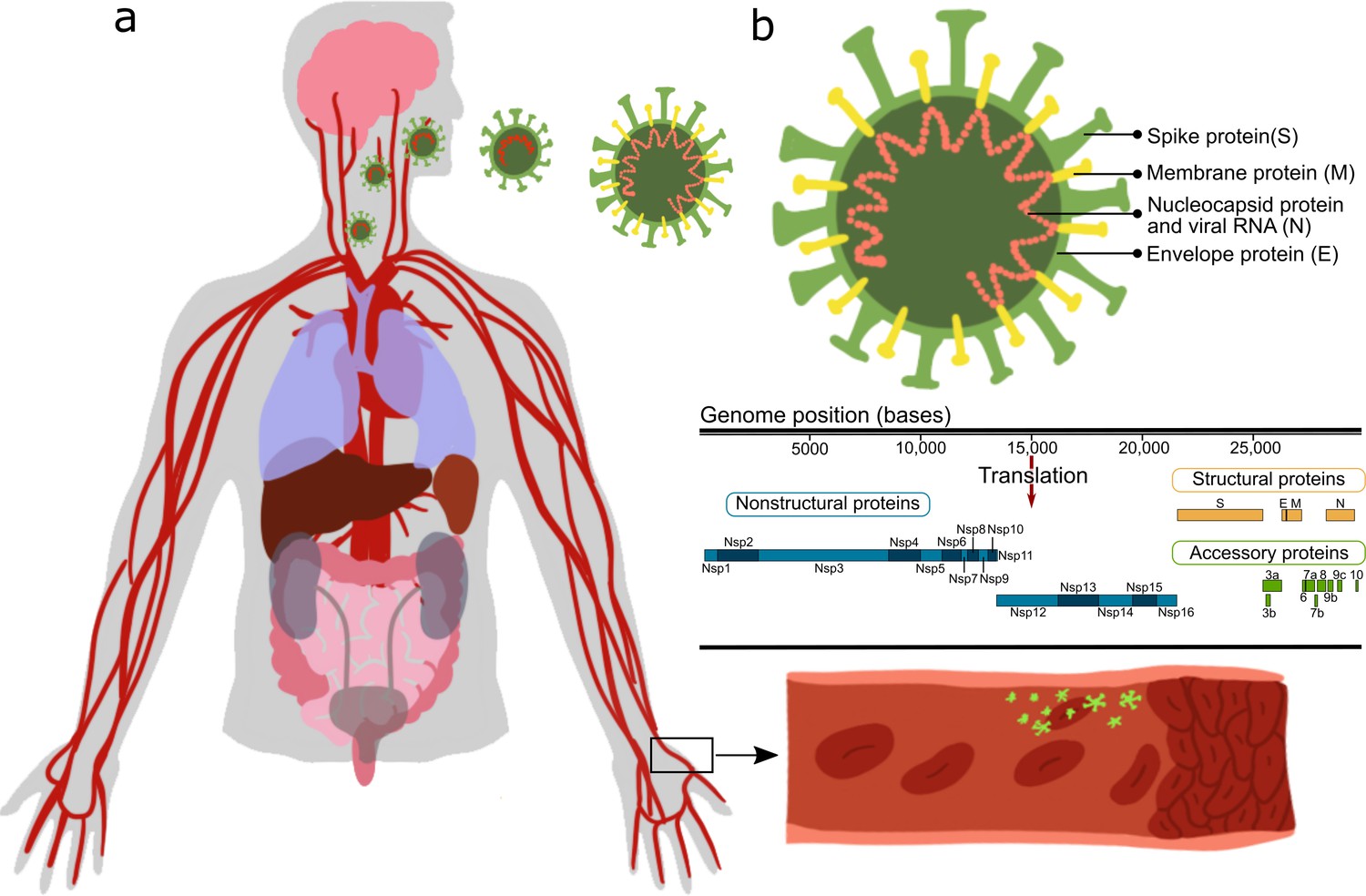
Table 1 – Severe acute respiratory syndrome (SARS)-CoV-2 proteins.
| SARS-CoV-2 proteins | General impact |
|---|---|
| Structural proteins | |
| S (spike) | Spike protein, mediates binding to ACE2, fusion with host membrane Surface glycoprotein, needs to be processed by cellular protease TMPRSS2 (Gordon et al., 2020) |
| M (membrane) | Membrane glycoprotein, the predominant component of the envelope A major driver for virus assembly and budding (Gordon et al., 2020) |
| E (envelope) | Envelope protein, involved in virus morphogenesis and assembly Coexpression of M and E is sufficient for virus-like particle formation and release (Gordon et al., 2020) |
| N (nucleocapsid) | Nucleocapsid phosphoprotein binds to RNA genome (Gordon et al., 2020) |
| Nonstructural proteins | |
| nsp1 | Leader sequence, suppresses host antiviral response Antagonizes interferon induction to suppress host antiviral response (Gordon et al., 2020) |
| nsp2 | Interferes with host cell signaling, including cell cycle, cell-death pathways, and cell differentiation May serve as an adaptor for nsp3 Not essential for virus replication, but deletion of nsp2 diminishes viral growth and RNA synthesis (Gordon et al., 2020; Procko, 2020) |
| nsp3 | nsp3–nsp4–nsp6 complex involved in viral replication Functions as papain-like protease (Gordon et al., 2020) |
| nsp4 | nsp3–nsp4–nsp6 complex involved in viral replication (Gordon et al., 2020) The complex is predicted to nucleate and anchor viral replication complexes on double-membrane vesicles in the cytoplasm (mitochondria) |
| nsp5 | Inhibits interferon I signaling processes by intervening in the NF-κB process and breaking down STAT one transcription factor Functions as 3-chymotrypsin-like protease, cleaves the viral polyprotein (Gordon et al., 2020) |
| nsp5_c145a | Catalytic dead mutant of nsp5 (Gordon et al., 2020) |
| nsp6 | nsp3–nsp4–nsp6 complex involved in viral replication Limits autophagosome expansion Components of the mitochondrial complex V (the complex regenerates ATP from ADP) copurify with nsp6 (Gordon et al., 2020) |
| nsp7 | Cofactor of nsp12 nsp7–nsp8 complex in part of RNA polymerase (nsp7, 8, 12 – replication complex)Affects electron transport, GPCR signaling, and membrane trafficking (Gordon et al., 2020; Peng et al., 2020b; Romano et al., 2020; Hillen et al., 2020) |
| nsp8 | Cofactor of nsp12 nsp7–nsp8 complex in part of RNA polymerase. Affects the signal recognition particle and mitochondrial ribosome (Gordon et al., 2020; Peng et al., 2020b; Romano et al., 2020; Chi et al., 2003) |
| nsp9 | ssRNA binding protein (can bind both DNA and RNA, but prefers ssRNA) Interacts with the replication complex (nsp7, 8, 12) (Cornillez-Ty et al., 2009) |
| nsp10 | Cofactor of nsp16 and nsp14 (Romano et al., 2020) Essential for nsp16 methyltransferase activity (stimulator of nsp16) Zinc finger protein essential for replication (Gordon et al., 2020; Peng et al., 2020b) |
| nsp11 | Unknown function |
| nsp12 | Functions as an RNA-direct RNA polymerase, the catalytic subunitAffects the spliceosome (Gordon et al., 2020; Peng et al., 2020b; Romano et al., 2020; Hillen et al., 2020) |
| nsp13 | Has helicase and 5’ triphosphatase activity Initiates the first step in viral mRNA capping nsp13,14,16 installs the cap structure onto viral mRNA in the cytoplasm instead of in the nucleus, where the host mRNA is capped (Gordon et al., 2020; Peng et al., 2020b; Romano et al., 2020; Ivanov et al., 2004) |
| nsp14 | In addition to the capping function of the methyltransferase, nsp14 is also an endonuclease (3’–5’ exoribonuclease) that corrects mutations during genome replication (Gordon et al., 2020; Peng et al., 2020b; Romano et al., 2020) |
| nsp15 | Endoribonuclease has uridine-specific endonuclease activity, essential for viral RNA synthesis (Gordon et al., 2020; Romano et al., 2020) |
| nsp16 | May involve complexation with nsp10 and nsp14, for stabilization of homoenzyme, for capping the mRNA (Gordon et al., 2020; Peng et al., 2020b; Romano et al., 2020) |
| Open reading frame (accessory factors) | |
| orf3a | Packaging into virions Mediates trafficking of spike protein by providing ER/golgi retention signals Induces IL-6b, activates NF-κB, activates the NLRP3 inflammasome (Gordon et al., 2020) |
| orf3b | Interferon antagonist and involved in pathogenesis (Gordon et al., 2020) |
| orf6 | Type I interferon antagonist, suppresses the induction of interferon, and interferon signaling pathways (Gordon et al., 2020) |
| orf7a | May be related to viral-induced apoptosis (Gordon et al., 2020) |
| orf7b | Unknown function |
| orf8 | Recombination hotspot Induces ER stress and activates NLRP3 inflammasomes Low similarity to SAR-CoV (Gordon et al., 2020) |
| orf9b | Suppresses host antiviral response Targets the mitochondrion-associated adaptor molecules MAVS and limits host cell interferon responses (Gordon et al., 2020) |
| orf9c | No evidence that this protein is expressed during SARS-CoV-2 infection (Gordon et al., 2020) |
| orf10 | No evidence that this protein is expressed during SARS-CoV-2 infection (Gordon et al., 2020) |
To tackle these challenges, we cultured human umbilical vein endothelial cells (HUVECs) and systematically transduced them with lentiviral particles encoding 26 out of the 29 viral proteins, separately. The three remaining genes were not included in this study purely for technical reasons. We then examined their effects on HUVEC monolayer permeability and the expression of factors involved in vascular permeability and coagulation.
The results were analyzed in the context of virus–host and host–host PPI networks. By combining the insights from the experimental and computational results, we generated a model that explains how each of the 26 proteins of SARS-CoV-2, including a mutated form of nsp5, the catalytic dead mutant termed nsp5_c145a, affects the protein network regulating vascular functionality. Moreover, once the PPI model was validated with our experimental data, we applied it to more than 250 proteins that have been identified in the literature as affected by the SARS-CoV-2 proteins.
This enabled us to pinpoint the more dominant SARS-CoV-2 proteins and chart their effects. Overall, this work shows how each of the SARS-CoV-2 proteins differentially affects vascular functionality; moreover, once the model was validated, we applied it to identify how SARS-CoV-2 proteins interact with proteins that have been significantly correlated with changes in cell functionality.



{kind=link}