










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



The clinical spectrum of the SARS-CoV-2 infection appear to be wide, encompassing asymptomatic infection, mild upper respiratory tract illness, and severe viral pneumonia with respiratory failure and death. Although COVID-19 cardinally manifests as diffuse alveolar damage, the involvement of other organ systems merits discussion.
Recent literature suggests that the kidney is a vulnerable organ in COVID-19 patients with evidence of acute kidney injury (AKI) in up to 37.5% of fatal cases of COVID-19 [5]. Despite the recognition of AKI as a frequent complication of COVID-19, there is a lack of consensus data regarding its incidence and outcomes. This systematic literature review aims to bridge this knowledge gap and help establish widespread consensus using worldwide data.
There is a dearth of accurate consensus regarding incidence, pathogenesis, diagnosis, management, and outcomes of AKI. This systematic literature review and meta-analysis specifically aims to address incidence and outcomes of AKI in COVID-19 patients.
Our findings revealed that AKI was a frequently observed complication of COVID-19 infection, with a cumulative incidence of 19.76%. COVID-19 patients had an overall mortality rate of 17.51%. This is in line with the historical mortality estimates of the SERS and MERS epidemics (10 and 40%, respectively) [65, 66].
COVID-19 patients with AKI had a significantly higher mortality rate of 54.24% and, overall, 18 times higher risk of death when compared to COVID-19 patients without AKI.
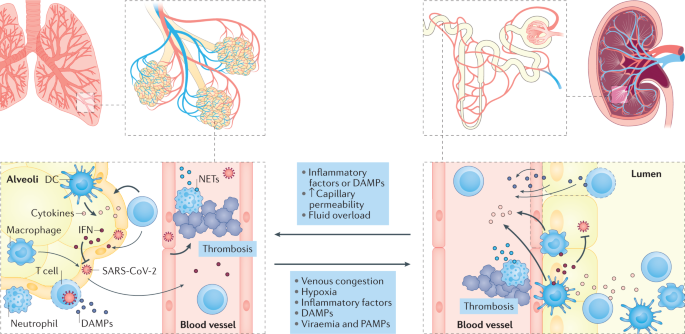
The pathophysiology of AKI in COVID-19 is complex, suggestive of a biphasic pattern with volume responsive AKI occurring in early stages of the disease and kidney injury occurring in later stages of the disease [67]. Acute lung injury that ensues in COVID-19 patients can effectuate impairment of the kidney function through its effect on gas exchange, release of pro-inflammatory mediators (IL-2, IL-7, IL-10, IFN-γ, IP10, GCSF, and MCP1), and cardiopulmonary interactions [67, 68].
The lung-kidney crosstalk may be bidirectionally deleterious, due to a cascade of irreversible cytokine release [67, 69]. A transient febrile/illness-related proteinuria may also be seen, like the MERS CoV infection [6, 70]. The cellular entry of SARS-CoV-2 is mediated by angiotensin-converting enzyme II, which is highly expressed in the podocytes and proximal tubules (100× levels compared to the lungs) and upregulated in patients with COVID-19 [7, 71-73].
An autopsy series conducted by Puelles et al. [74] (n = 27), showed that SARS-CoV-2 has organotropism beyond the pulmonary tract. Recent postmortem kidney pathology findings in patients with COVID-19 suggests that although there are little signs of viral nephropathy, noted acute tubular injury in the setting of creatinine elevation is reversible with appropriate care such as aggressive fluid management [75].
Mechanical ventilation may be provided to patients which affects the renal and systemic hemodynamics, stimulates sympathetic pathways, and causes biotrauma [9, 76].
In terms of management of patients with COVID-19 and AKI, discussion of goals of supportive care (e.g., KRT) among clinicians and patients/families is recommended. Standard recommendations by KDIGO include nutritional and fluid support, avoidance of nephrotoxins, maintenance of oxygenation saturation, and hemodynamic stability for the management of AKI. KRT may reduce the burden of inflammatory mediators and cytokines as supported by a study with Ghani et al. [77-80] particularly to patients with AKI; however, more studies are required.
Various modalities such as peritoneal dialysis, hemodialysis, CKRT, and sustained low-efficiency dialysis can be utilized for AKI management. Although CKRT is the preferred modality in most ICU settings, the choice of modality should be based on the hemodynamic status, patient characteristics and needs, available resources, and health-care expertise [81-83].
Newer modalities such as CytoSorb (an extracorporeal cytokine filter) and Oxiris (a blood purification device for cytokine clearance) have received expedited FDA approval for use in adult COVID-19 patients (≥18 years old) with AKI [84, 85], where the provider can obtain an exploratory investigational new drug for use in the pediatric population. Evidence is actively evolving but insufficient for widespread application in COVID-19 patients with AKI.
The mechanism of how this was occurring was poorly understood for a long time.
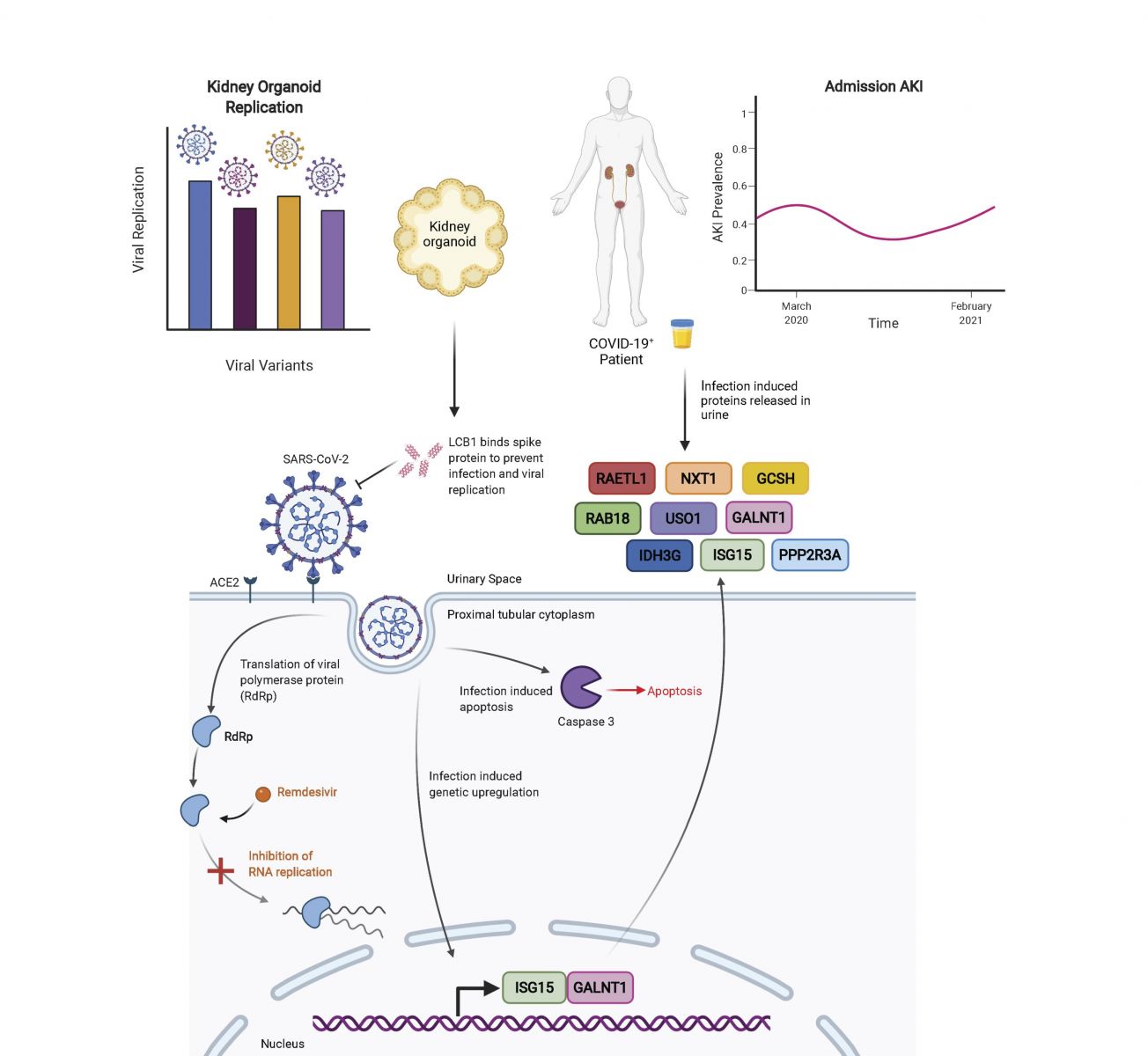
This new study by researchers from the University of Washington School of Medicine confirms that that SARS-CoV-2 can directly invade human kidney cells causing a range of kidney issues including acute kidney injury!
Graphical Abstract
Kidneys are critical target organs of COVID-19, but susceptibility and responses to infection remain poorly understood.
The study team introduced SARS-CoV-2 variants with genome edited kidney organoids and utilized clinical data to investigate tropism, mechanism, and therapeutics, findings alarmingly showed that SARS-CoV-2 specifically infects organoid proximal tubules amongst diverse cell types. Infections produce replicating virus, apoptosis, and disrupted cell morphology, features of which are revealed in the context of polycystic kidney disease.
Cross-validation of gene expression patterns in organoids reflect proteomic signatures of COVID-19 in the urine of critically ill patients indicating interferon pathway upregulation. SARS-CoV-2 viral variants Alpha, Beta, Gamma, Kappa, and Delta exhibit comparable levels of replication in organoids. Infection is ameliorated in ACE2-/- organoids and blocked via treatment with de novo designed spike binder peptides.
The study findings collectively clarify the impact of kidney infection in COVID-19 as reflected in organoids and clinical populations, enabling assessment of viral fitness and emerging therapies.
The research were published in the peer reviewed journal: JCI Insight
https://insight.jci.org/articles/view/154882
Other referece …
https://annalsofintensivecare.springeropen.com/articles/10.1186/s13613-021-00914-5
https://www.nature.com/articles/s41581-021-00452-0
Pathophysiology of COVID-19 AKI
The pathophysiology of COVID-19 AKI is thought to involve local and systemic inflammatory and immune responses, endothelial injury and activation of coagulation pathways and the renin–angiotensin system31,35. Direct viral infection with renal tropism of the virus has also been proposed but remains controversial36. Non-specific factors that are common in critically ill patients, such as mechanical ventilation, hypoxia, hypotension, low cardiac output and nephrotoxic agents, might also contribute to kidney injury and/or functional decline in the most severely affected patients (Box 1).
Box 1 Factors that may contribute to COVID-19-associated acute kidney injury
Acute tubular injury
- Regional inflammation
- Direct viral infection
- Renal compartment syndrome
- Tissue hypoxia hypoperfusion leading to hypoxaemia, hypotension, hypovolaemia and heart failure
- Nephrotoxic-induced injury (potentially associated with the use of antibiotics (vancomycin, aminoglycosides, colistin) or antivirals (remdesivir, ritonavir))
- Rhabdomyolysis
Vascular injury
- Endotheliitis
- Microthrombi
- Thrombotic microangiopathy
Glomerular injury
- Collapsing glomerulopathy (potentially caused by interferon-associated podocyte injury)
- Glomerulonephritis
Interstitial injury
- Acute interstitial nephritis; infiltration by immune cells
- Interstitial oedema
COVID-19, coronavirus disease 2019.
Insights from renal histology
Autopsy studies demonstrate that acute tubular injury is by far the most common finding in kidneys of patients with COVID-19 AKI (Supplementary Table 1). Of note, tubular autolysis is a confounding factor in post-mortem histological analyses of acute tubular injury31,37.
Analyses of post-mortem kidney samples from patients with stage 2 or 3 AKI and COVID-19 have revealed acute tubular injury characterized by mostly mild focal acute tubular necrosis29,33,35,38, illustrating an apparent uncoupling between the extent of histological injury and decline of kidney function — a finding previously reported in patients with non-COVID sepsis39. In an autopsy series of 9 patients in the UK, evidence of acute tubular injury was noted in all patients; viral load quantified by the use of quantitative real-time PCR targeting the viral E gene was observed in the kidneys of 3 patients and detection of subgenomic viral RNA in only 1 (11%) kidney sample38,40.
Another analysis of kidney biopsy samples from 17 patients with SARS-CoV-2 infection and mostly mild COVID-19 symptoms identified AKI and proteinuria in 15 and 11 patients, respectively. Acute tubular injury (n = 14; 82%), collapsing glomerulopathy (n = 7; 41%) and endothelial injury or thrombotic microangiopathy (n = 6; 35%) were the most common histological findings41 (Supplementary Table 1). Virus detection (using immunohistochemistry for SARS-CoV-2 nucleocapsid and RNA in situ hybridization) were negative in patient samples on which it was performed.
Another series from France demonstrated tubular injury in the most severely ill cohort whereas glomerular pathology was restricted to the non-ICU patients34. Of note, most biopsies were performed several weeks after the onset of COVID-19 symptoms, and most have failed to show notable SARS-CoV-2 infection of the kidney. Despite initial concerns about the methodology and interpretation of some early studies that reported direct viral tropism of the kidney36,42,43,44, one study that identified and isolated SARS-CoV-2 from post-mortem kidney tissue demonstrated that the virus could replicate in non-human primate kidney tubular epithelial cells, showing its ability to infect kidney cells45.
The researchers further identified that 23 of 32 patients with AKI (72%) showed viral RNA in kidney tissue, whereas viral RNA was identified in only 3 of 7 (43%) patients without AKI. Another autopsy study that performed microdissection of kidneys from 6 patients with COVID-19 identified SARS-CoV-2 in different kidney compartments, particularly in the glomerulus43. Viral RNA and protein were also detected in kidney by in situ hybridization with confocal microscopy.
In addition, SARS-CoV-2 particles have been observed in urine samples33,46,47 — a finding that either reflects the release of virus from infected, damaged tubule epithelial cells or the filtration of viral fragments, as the high molecular weight of SARS-CoV-2 (600 kDa) should prevent it from being filtered through the intact glomerular filtration barrier48. Thus, a substantial body of evidence now suggests that SARS-CoV-2 can infect kidney tissue; however, a direct role of the virus in the development of AKI remains to be confirmed.
Collapsing glomerulopathy
Collapsing glomerulopathy has been reported in several patients with COVID-19 (Supplementary Table 1). This entity has been described as COVID-19-associated nephropathy (COVAN), and seems to occur mostly in patients with non-severe respiratory symptoms of COVID-19 and isolated AKI or in those presenting with glomerular proteinuria30,32,34. Of note, collapsing glomerulopathy has previously been described in the context of other viral infections, including HIV parvovirus B19, cytomegalovirus and Epstein–Barr virus infections.
COVAN is associated with high-risk APOL1 genotypes, and has been observed mostly in Black patients. The true incidence of collapsing glomerulopathy and its contribution to kidney failure in the context of COVID-19 compared with the effects of other underlying conditions (for example, hypertension or CKD) is unknown. Although the exact pathophysiology of COVAN remains unknown, it may share common mechanisms with HIV-associated nephropathy, with podocyte injury through disruption of autophagy and mitochondrial homeostasis31.
Endothelial dysfunction and coagulation
Biomarkers of coagulation and fibrinolysis activation (for example, fibrinogen and D-dimer) have been repeatedly associated with an increased risk of death in patients with COVID-19. Autopsy studies have reported a ninefold higher incidence of observed microvascular and macrovascular thrombosis in lungs of patients with COVID-19 than that of patients with influenza pneumonia49. Systemic microvascular and macrovascular thrombosis in organs, including the kidneys, have also been repeatedly reported in the context of COVID-19 (refs50,51,52).
Many critical illnesses are associated with microvascular and endothelial injury but SARS-CoV-2 is believed to specifically affect the endothelium. Post-mortem studies have reported vascular endotheliitis in patients with COVID-19 (refs49,53). Moreover, findings from at least one report indicate viral infection of kidney endothelial cells53; however, that report used electronic microscopy to identify viral elements, which is insufficiently specific and thus firm evidence of direct viral infection of kidney endothelial cells is lacking.
Nonetheless, increased levels of plasma biomarkers of endothelial injury (for example, soluble (s) E-selectin, sP-selectin, ANG2, sICAM1 and von Willebrand factor antigen) and platelet activation (soluble thrombomodulin) are associated with poor prognosis54,55,56. Microvascular inflammation can trigger endothelial activation, leading to vasodilation, increased vascular permeability and pro-thrombotic conditions57,58,59.
Complement activation — evidenced by increased circulating levels of soluble complement components C5b–9 and C5a and by tissue deposition of C5b–9 and C4d in lung and kidney tissues60,61,62 — may further promote inflammation and coagulation pathways in COVID-19. The release of damage-associated molecular patterns from cells undergoing necrosis might further contribute to endothelial injury in COVID-19 (ref.63). SARS-CoV-2 has furthermore been shown to bind to platelets via ACE2, leading to platelet activation and immunothrombosis64,65,66.
Thus, platelet activation may represent a potential player in the pathophysiology of COVID-19 AKI67,68. Circulating prothrombotic autoantibodies that target phospholipids and phospholipid-binding proteins have also been reported69. In a cohort of 172 hospitalized patients with COVID-19, higher titres of prothrombotic antibodies were associated with lower eGFR. In vitro studies confirmed the autoantibodies to be drivers of endothelial cell activation, potentially contributing to the thrombo-inflammatory effects observed in severe COVID-19 (ref.70).
However, macrothrombi and microthrombi have been inconsistently observed in kidneys of patients who have died with COVID-19, or have involved only a small proportion of renal capillaries. A small autopsy study from New York, USA, observed thrombotic microangiopathy within glomeruli in only 1 of 7 cases51.
Another series of kidney biopsy samples from 17 patients with mild COVID-19 symptoms identified evidence of acute glomerular endothelial cell injury in 6 patients, most of whom demonstrated laboratory features of thrombotic microangiopathy41. Of note, no evidence of peritubular vascular injury was observed in that study. Neutrophils and neutrophil extracellular traps — frequently aggregating with platelets — have been observed in many organs including the kidneys, despite the sporadic presence of virus on histology, suggesting a role of inflammation in the development of intravascular thrombi71. Cases of renal artery thrombosis have also been anecdotally reported72,73. Finally, patients with severe COVID-19 often present with complications associated with chronic endothelial dysfunction, such as hypertension or diabetes, which are themselves associated with decreased endothelial nitric oxide synthase activity and bioavailability of nitric oxide — a major vasodilator and antithrombotic factor74.
The immune and inflammatory response
Several alterations in both innate and adaptive immune responses have been reported following SARS-CoV-2 infection. Immunosenescence — a term used to define the innate and adaptive immunological alterations that occur with ageing — is characterized by inflammageing, a low-grade inflammatory state that may have a key role in determining organ dysfunction, as well as by ineffective T cell responses and antibody production. These features have been reported in COVID-19 and may in part explain the higher mortality among elderly individuals and those with comorbidities as a consequence of inefficient viral clearance, the enhanced release of cytokines and chemokines, endothelial damage and activation of the coagulation and complement cascades75.
Inflammation
The enhanced release of inflammatory mediators by immune and resident kidney cells is likely to be a key mechanism of tissue damage in patients with COVID-19. Inflammatory mediators, such as TNF and FAS, can bind to their specific receptors expressed by renal endothelial and tubule epithelial cells causing a direct injury76,77. Such interactions have been observed in experimental models of sepsis and are supported by measurements of plasma cytokine levels in patients with sepsis-associated AKI78, although their role in COVID-19 AKI is yet to be clearly demonstrated.
Interferon
Other studies have demonstrated a key role for type I interferon responses in suppressing viral replication and regulating the immune response in the context of COVID-19. Available evidence suggests that SARS-CoV-2 infection can lead to suppression of interferon release; moreover, patients treated with interferon demonstrated improved viral clearance with a concomitant reduction in levels of IL-6 and C-reactive protein79. One study demonstrated that patients with inborn errors of type I interferon immunity and extremely low serum levels of IFNα (<1 pg/ml) are at a greater risk of severe COVID-19 than those with higher IFNα levels (1–60 pg/ml))80.
The same group of researchers also identified individuals with severe COVID-19 with autoantibodies directed against type I interferon, suggesting a possible autoimmune basis to the inefficient blockade of SARS-CoV-2 infection as a result of low interferon plasma levels81. These findings justify the ongoing clinical trials of therapeutic interferon administration for patients with COVID-19 (refs82,83).
However, a note of caution is warranted given that interferons are well-known mediators of glomerular injury. Indeed, IFNα and IFNβ exert differential effects on parietal epithelial cells and podocytes, acting to enhance podocyte loss and promote glomerulosclerosis, respectively84. Moreover, proteinuria occurring in the context of inflammation has been ascribed to podocyte injury following cytokine release and the activation of type I interferon signalling85. Finally, APOL1 risk alleles may promote glomerular damage via a process that involves interferon86.



{kind=link}