










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")


A new detailed international study of the genome of the Omicron variant and its mutations and location of mutation clusters has revealed that the Omicron’s Spike protein structure has accommodated significant sequence changes, likely in response to selective pressures favoring increased transmission, immune evasion, or viral replication-either at the population level or in a single or group of chronically infected individuals and has potentially acquired new functionality.
The study findings were published on the Virological.org site.
https://virological.org/t/selection-analysis-identifies-significant-mutational-changes-in-omicron-that-are-likely-to-influence-both-antibody-neutralization-and-spike-function-part-1-of-2/771
https://virological.org/t/selection-analysis-identifies-significant-mutational-changes-in-omicron-that-are-likely-to-influence-both-antibody-neutralization-and-spike-function-part-2-of-2/772
Three possible explanations for the missing intermediates are: (1) SARS-CoV-2 sampling in Southern Africa between May and September 2021 might have been too sparse or biased to detect low frequency variants amongst high numbers of Delta variant infections during this time period; (2) long-term evolution in one or more chronically infected people – similar to the proposed origin of lineages such as Alpha and C.1.2 (1) 5 (2) 2 (3) 1 – where intermediate forms would have remained unsampled within the individual(s); and (3) a reverse zoonosis to a non-human host, followed by undetected spread therein, and a spillover back into humans.
Regardless of the route that Omicron took to eventual community transmission, its genome accumulated 53 mutations relative to the Wuhan-Hu-1 reference strain, with 30 non-synonymous substitutions in the Spike encoding S gene alone.
Here, we characterize the selective pressures that may have acted during the genesis of the Omicron variant and curate available phenotypic and genomic variation data associated with Omicron mutations.
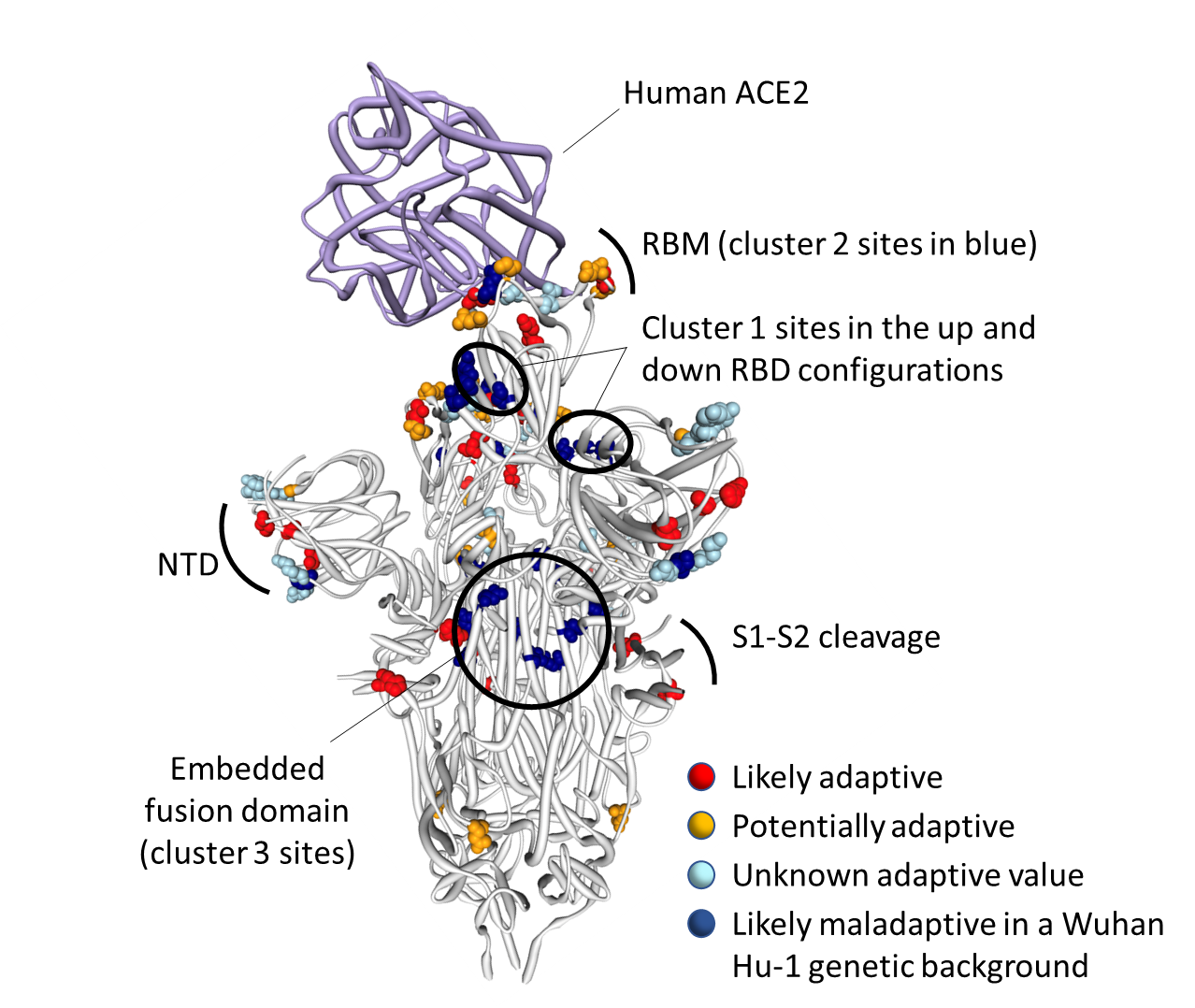
We use these comparisons to identify which Omicron mutations might contribute to transmission advantages, immune escape, or novel spike functionality. Our analysis identifies three clustered sets of mutations in the Spike protein, involving 13 amino acids that have previously been highly conserved across SARS-CoV-2 and other Sarbecoviruses.
This dramatic about-face in evolutionary dynamics at these 13 sites suggests that Omicron’s Spike protein structure has accommodated significant sequence change, likely in response to selective pressures favoring increased transmission,immune evasion, or viral replication – either at the population level or in a single or group of chronically infected individuals—and has potentially acquired new functionality.
Many of the Omicron S-gene mutations are likely contributors to viral adaptation
For context, this fraction of positively selected sites (0.53) is approximately four times higher than the fraction of all SARS-CoV-2 S-gene sites that have ever shown any signals of positive selection (0.14).



{kind=link}