










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Rosemary contains carnosic acid (CA) and carnosol (CS), abietane-type phenolic diterpenes, which account for most of its biological and pharmacological actions, although claims have also been made for contributions of another constituent, rosmarinic acid.
This review focuses on the potential applications of CA and CS for Alzheimer’s disease (AD), Parkinson’s disease (PD), and coronavirus disease 2019 (COVID-19), in part via inhibition of the NLRP3 inflammasome.
CA exerts antioxidant, anti-inflammatory, and neuroprotective effects via phase 2 enzyme induction initiated by activation of the KEAP1/NRF2 transcriptional pathway, which in turn attenuates NLRP3 activation. In addition, we propose that CA-related compounds may serve as therapeutics against the brain-related after-effects of SARS-CoV-2 infection, termed “long-COVID.”
Many reports hold that unregulated NLRP3 inflammasome activation may potentially contribute to the severity of COVID-19 and its aftermath. It is therefore possible that suppression of NLRP3 inflammasome activity may prove efficacious against both acute lung disease and chronic neurological after-effects.
Because CA has been shown to not only act systemically but also to penetrate the blood–brain barrier and reach the brain parenchyma to exert neuroprotective effects, we discuss the evidence that CA or rosemary extracts containing CA may represent an effective countermeasure against both acute and chronic pathological events initiated by SARS-CoV-2 infection as well as other chronic neurodegenerative diseases including AD and PD.
Anti-Inflammatory Actions of Rosemary and Carnosic Acid
In this study, we present both an overview and new primary data describing the therapeutic effects of the herb rosemary, concentrating on its primary medicinal constituent, carnosic acid (CA). We focused on the effects of CA on neurological disorders including Alzheimer’s disease (AD), Parkinson’s disease (PD), and infection with SARS-CoV-2 causing coronavirus disease 2019 (COVID-19), as well as the long-term consequences of the infection, termed long-COVID.
Uses of Rosemary and Rosemary Extract
Rosemary is an economically important plant species that is widely distributed due to its culinary, medicinal, and cosmetic uses [1,2,3]. Both fresh and dried leaves of rosemary have been used for their characteristic aroma and flavor in many food dishes. Additionally, rosemary extracts have been employed as lipophilic antioxidants to preserve foods against oxidation from environmental stress [1,2,3].
Because of these characteristics, the European Union, United States, and Japan have all approved rosemary extracts as food additives for food preservation [4,5,6]. Rosemary contains two groups of potentially active compounds. One group is comprised of small molecular weight aromatic compounds, called “essential oils”, which rapidly evaporate to produce the characteristic smell and taste of rosemary.
The other group is represented by the polyphenolic compounds including carnosic acid (CA) and carnosol (CS), which have been shown to manifest direct and indirect antioxidant actions [7,8,9]. While our group has focused on the effects of CA, we also report here the work of others on CS.
Anti-Inflammatory Effects of CA on Tissue Macrophages
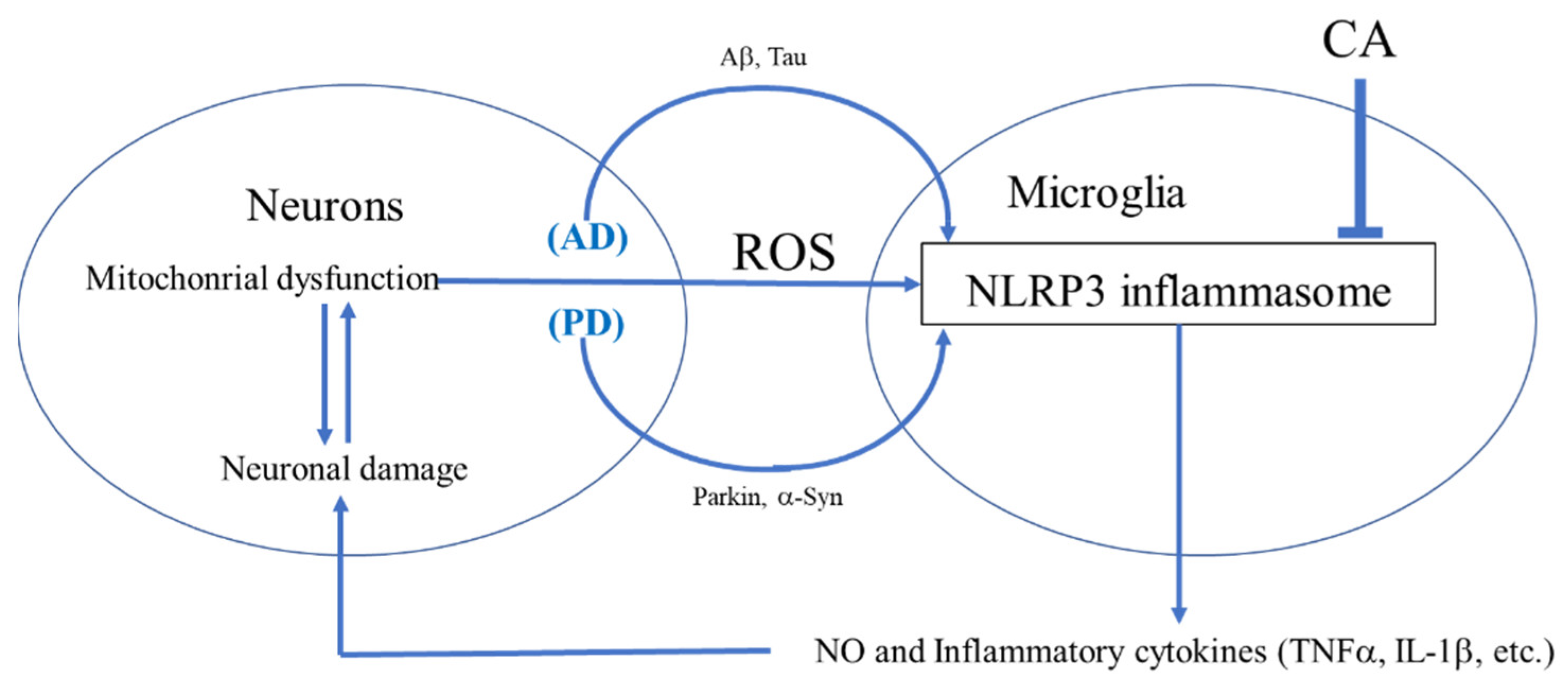
While CA may act on a variety of cell types, we concentrated here on monocytoid cells of the innate immune system. CA-mediated modulation of the macrophage and microglial inflammatory response in AD and PD via inhibition of NLRP3 could potentially have significant therapeutic value (Figure 1) [10,11].
Along these lines, studies have revealed that rosemary extract or CA can inhibit inflammatory cytokine expression (i.e., tumor necrosis factor-α (TNF-α)), in Raw 264.7 macrophage cells [10,11]. CA also attenuates LPS-induced nitric oxide (NO)/reactive nitrogen species (RNS) production in these macrophage cells [11].
Furthermore, CA can suppress the TNF-α signaling pathway by inhibiting the inhibitor of nuclear factor κ-B (NF-κB) as well as via upregulation of HO-1 expression [12,13]. As discussed in more detail below, by upregulating erythroid derived 2-related factor 2 (NRF2) transcriptional activity, CA leads to the downregulation of the TNF-α and NO inflammatory response [14,15,16,17].
Moreover, in vivo, CA has also been shown to inhibit the LPS-induced rise in serum levels of proinflammatory cytokines including TNF-α and interleukin (IL)-6; many of these effects of CA are due to NRF2 activation [14]. For these in vivo studies, CA was suspended in 0.5% solution and administered (100 mg/kg) by gavage once daily for three days. Activation of NRF2 was confirmed by NRF2 nuclear translocation and increased expression of antioxidant genes known to be transcriptionally regulated by the NRF2 pathway [14].
Anti-Inflammatory Effects of CA on Brain Microglia
Microglial cells are the major resident innate immune inflammatory cells of the brain and, upon activation, can produce proinflammatory cytokines (e.g., IL-1β, IL-6, and TNF-α). In an in vitro model of brain infection, CA was found to inhibit LPS-induced activation of mouse microglia, thus decreasing the release of inflammatory cytokines such as IL-1β and IL-6 [15,16,17].
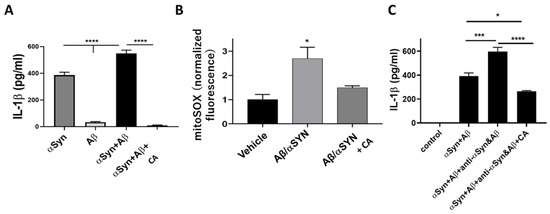
CA is also reported to decrease NO production associated with inducible NO synthase [15,16,17]. Several studies have observed that misfolded proteins such as α-synuclein (α-Syn), particularly in combination with amyloid-β (Aβ) or possibly Tau and release of inflammatory cytokines from microglia is critical for microglial–astrocyte–neuron signaling in neurodegenerative disorders (Figure 2) [18,19,20]. In addition, microglial-mediated inflammation is reportedly upregulated in human AD and PD brains [18,19,20]. Based on various inflammatory models, the therapeutic potential of CA has been suggested for a number of neurodegenerative disorders including AD and PD [18,19,20].
Complicated Manifestations of COVID-19
Manifestations
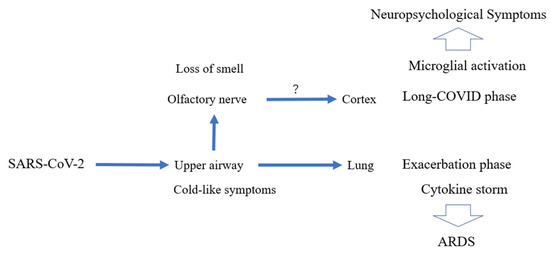
COVID-19, caused by the virus SARS-CoV-2, has rapidly become the most urgent issue in global public health [73,74]. OVID-19 is composed of many complicated symptoms in addition to acute respiratory distress syndrome (ARDS) as well as physiological and biochemical alterations (Figure 5) [73,74].
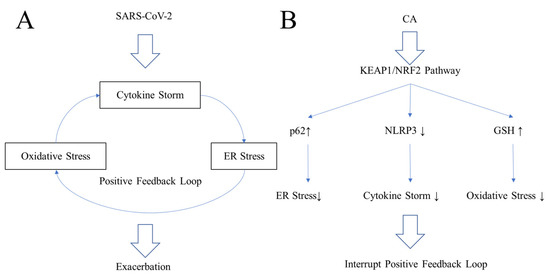
These include the translocation of T cells into the lung, alterations in coagulation, and increased ferritin levels, a mediator of immune dysregulation [75,76]. Contributing to these manifestations is a positive feedback loop at the cellular level composed of cytokine storm, oxidative stress, and ER stress (Figure 5) [75,76]. Since few specific therapies (other than vaccination) are available, patients have been treated symptomatically with oxygen and broad-spectrum antiviral drugs such as interferon-α and glucocorticoids [75,76].
Remdesivir, a medicine used to treat Middle East Respiratory Syndrome (MERS), is at least partly effective against COVID-19 infection [75,76]. More specific therapeutic drugs, such as Pfizer’s Paxlovid and Merck’s molnupiravir, are being developed and have just been released as of this writing to supplement widespread vaccination efforts [75,76].
Cytokine Storm
Initially, SARS-CoV-2 infects epithelial cells of the upper respiratory tract. If the infection is limited to these cells, the disease can manifest mild symptoms (Figure 6) [77,78]. Severe inflammation develops when the virus enters the lungs [77,78]. Translocation of T lymphocytes into the alveoli may contribute to inflammation, leading to the development of ARDS [77,78]. In severe cases, the virus can enter the blood circulation and spread the infection to other parts of the body (Figure 6) [77,78]. The “damage-associated molecular patterns (DAMPs)” released from the dead cells activate immune cells and can induce cytokine storm [77,78]. Thus, in severe forms of COVID-19, hyperactivation of the immune system can contribute to multiple organ failure [77,78]. In general, tightly controlled activation of the innate immune system is essential for viral recognition and clearance of viruses [77,78]. However, dysregulated inflammation causes a cytokine storm due, in part, to NLRP3 inflammasome activation [79,80]. Cytokine storm can also contribute to hypercoagulation in microvessels, another component of multi-organ failure [79,80]. Patients in critical condition often manifest systemic inflammatory markers such as high levels of cytokines, including IL-6, TNF-α, and IL-8 [79,80]. Thus, measures to counter cytokine storm have been proposed as part of the treatment regimen [77,78,79,80].
Oxidative Stress in COVID-19
Inflammatory cytokines and ROS act together to activate lung epithelial cells [79,80]. This leads to dysregulated cell contact, greater cell permeability, and influx of fluid, collectively contributing to lower oxygen tension [77,78]. Oxidative stress and cytokine storm also lead to dysfunction and apoptosis of endothelial cells and activation of the coagulation system [81,82]. Accumulation of serum ferritin and Fe3+ have been reported in severe COVID-19 [81,82]. Ferritin can convert Fe3+ to Fe2+, which can then participate in the Fenton reaction [81,82], contributing further to oxygen free radical production. The increase in ROS can cause additional release of iron from stores with ferritin as well as damage to DNA, lipids, and proteins [81,82,83,84]. It has been proposed that hydrogen gas or high concentrations of vitamin C can at least partially prevent these events [83,84].
Endoplasmic Reticulum (ER) Stress in COVID-19
SARS-CoV-2 viral proteins expressed in infected cells are folded in the host cell endoplasmic reticulum (ER) [85,86]. Viral proteins are subject to modification in the intracellular membrane of the ER and contribute to ER stress [85,86]. Replication of SARS-CoV-2 requires a large number of proteins, and after multiple replication cycles, budding leads to viral release from the host cell [87,88].
Inhibiting the Positive Feedback Loop of COVID-19
SARS-CoV-2 induces a positive feedback loop consisting of cytokine storm, oxidative stress, and ER stress in lung epithelia during the exacerbation phase of COVID-19 (Figure 5A) [89,90]. Due to these complex features of COVID-19, classical anti-inflammatory drugs and antioxidants cannot fully restore lung function [89,90]. One countermeasure would be to disrupt the positive feedback loop (Figure 5B) [89,90]. To effectively combat the positive feedback loop that contributes to COVID-19 pathogenesis, NRF2 activators have been proposed as a potential therapeutic [90,91]. Until now, however, most proposed therapeutic interventions have focused on overcoming each stress individually rather than the complex positive feedback loop [81,82,83,84,85,86,87,88]. For example, dexamethasone is a steroid anti-inflammatory drug, while hydrogen gas or high concentrations of vitamin C are potential free radical scavengers [83,84]. However, none of these approaches completely suppresses ARDS. A more comprehensive approach to disrupting the positive feedback loop is probably required for an effective therapy (Figure 5B) [89,90]. Prior work has shown that NRF2 activators may protect the lung and brain from severe inflammation [89,90]. NRF2 can ameliorate inflammation by reducing IL-6 and IL-1β, in part by inhibiting activation of the NLRP3 inflammasome (Figure 5B) [89,90]. NRF2 induces the expression of genes encoding phase 2 antioxidant and anti-inflammatory enzymes, leading to an increase in GSH and preservation of redox homeostasis to protect against oxidative stress [36,37,38,39,40]. Additionally, NRF2 can induce the anchor protein, p62, which activates autophagy to ease ER stress [91,92]. Thus, NRF2 activators coordinately reduce ER stress, oxidative stress, and cytokine storm by inducing p62, inducing GSH synthetic enzymes, and inhibiting the NLRP3 inflammasome, leading to suppression of the positive feedback loop [89,90].
Potential Relationship of Neurological Sequelae in COVID-19 and Microglial Activation
Neurological symptoms induced by SARS-CoV-2 can produce cognitive symptoms called brain fog, the origin of which is still unclear. This symptom is common in what has been termed “long-COVID” [93,94]. Microglia, the innate immune cells of the CNS, are known to play a central role in the maintenance of brain homeostasis and inflammatory responses [93,94]. Normally, microglia can maintain homeostasis and contain inflammation, resulting in the improvement of symptoms [93,94]. However, during neurodegenerative disorders and possibly during COVID-19, the proinflammatory state of microglia could be deleterious. long-COVID patients persistently suffer from unacceptable neurological symptoms such as brain fog, fatigue, depression, and anxiety [93,94]. The question remains whether in long-COVID patients, microglia are hyperactivated and become proinflammatory, and if this process is accelerated by activation of the NLRP3 inflammasome [95]. If so, an inhibitor of the NLRP3 inflammasome could become an effective therapeutic for long-COVID.
CA as a Potential COVID-19 Therapeutic via Inhibition of NLRP3 Inflammasome, Other Inflammatory Pathways, and SARS-CoV-2 Infection
ARDS Mediated by NLRP3 Inflammasome
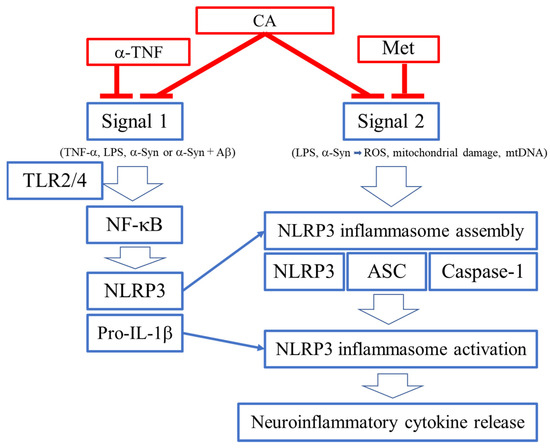
As schematically highlighted in Figure 7, ARDS is initiated by SARS-CoV-2 infection. Viral replication can lead to TLR signaling and NF-κB activation (Signal 1 in Figure 2) [96,97]. However, this signal itself is not the direct cause of ARDS [96,97]. It is the dysregulated, hyperinflammatory response to viral infection, mediated at least in part by NLRP3 inflammasome activation, which contributes to ARDS [96,97]. As reviewed above, the extreme inflammatory response is triggered by activation of the NLRP3 inflammasome and other inflammatory pathways that induce release of proinflammatory cytokines such as IL-1β, IL-18, IL-6, and TNF-α [32,33,34,35]. Thus, a potential countermeasure against the development of severe COVID-19 would be to prevent activation of NLRP3 and other inflammatory pathways [32,33,34,35].
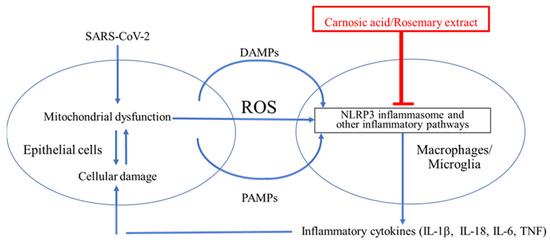
Figure 7.Cytokine storm contributes to ARDS in the lungs and possibly the brain of COVID-19 patients. SARS-CoV-2 binds to ACE2 and enters epithelial cells to induce ER stress due to overproduction of viral proteins [73,74,75,76]. The damaged cells release damage-associated molecular patterns (DAMPs), pathogen-associated molecular patterns (PAMPs), and ROS [24,25,26]. ROS, DAMPs, and PAMPs activate the NLRP3 inflammasome in surrounding macrophages in the lung and potentially in microglia in the brain [77,78]. This activation is critical for the release of inflammatory cytokines such as IL-1β from macrophages and microglia [77,78]. These cytokines enhance damage of epithelia and other cell types and may contribute to cytokine storm and the severe lung disease of ARDS [96,97]. The NLRP3 inflammasome is thus a plausible drug target for treating ARDS [77,78].
CA as a Potential Treatment for COVID-19 by Suppressing Infection and Cytokine Production
Recently, McCord et al., reported that PB125, a CA-based therapeutic agent, was potentially useful in the treatment of respiratory viral diseases, including COVID-19, in part by inhibiting cytokine storm [98,99,100]. They observed a marked downregulation of genes encoding inflammatory cytokines including IL-1β, IL-6, TNF-α, cell adhesion molecules (ICAM-1, VCAM-1, and E-selectin), and a group of interferon-γ-induced genes, suggesting that cytokine storm might be suppressed by CA [98,99,100]. McCord and colleagues further reported that PB125 downregulates ACE2 and TMPRSS2 mRNA expression in human liver-derived HepG2 cells, consistent with the notion that CA might inhibit viral entry, which ACE2 and TMPRSS2 proteins mediate [98,99,100]. These results suggest that the use of rosemary extract or CA might represent a possible therapeutic against acute ARDS in the lung and more protracted cytokine production [98,99,100]. Therefore, clinical testing of rosemary extract or CA for either acute COVID-19 lung disease or chronic (so-called long) COVID-19 might be considered. The rationale for such treatment is discussed further below. A caveat with these findings, however, is the fact that IL-1 or IL-6 inhibitors have been reported to suppress SARS-CoV-2 neutralizing antibodies in patients with COVID-19 [101]. Hence, the timing of any therapy aimed at suppressing these cytokines will have to be judiciously monitored in order not to interfere with anti-SARS-CoV-2 antibody therapies [98].
CA as a Potential Anti-Infectious Agent against SARS-CoV-2 by Inhibiting Binding to ACE2
Recently, two of us (C-k.O. and S.A.L.) found that CA can inhibit infection with SARS-CoV-2, approaching 90% efficacy, as assessed in a pseudovirus entry assay (Figure 8A). Mechanistically, CA, after conversion to the quinone form, is likely to S-alkylate critical thiol residue(s) on ACE2, the cell surface receptor for SARS-CoV-2 (Figure 8B). This reaction would then block the interaction of ACE2 with the Spike protein of SARS-CoV-2, and thus abrogate viral entry. Further work on reactions of these critical thiol groups in preventing SARS-CoV-2 infection is forthcoming.
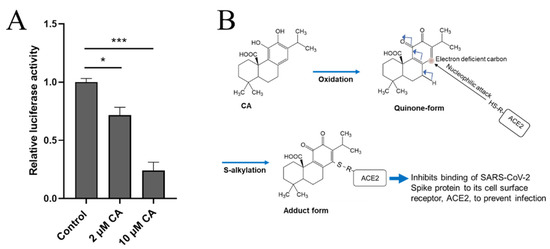
Figure 8.CA inhibits SARS-CoV-2 infection. (A) Inhibition of SARS-CoV-2 infection by CA in pseudovirus entry assay. HeLa cells stably-expressing ACE2 (HeLa-ACE2) were incubated with SARS-CoV-2 Spike (D614) pseudovirus particles in the presence and absence of the indicated concentration of CA, as described previously [102]. After 48 h, viral transduction efficiency was monitored by luciferase activity. Data are mean ± S.E.M., * p < 0.05, *** p < 0.001 by ANOVA with Tukey’s multiple comparisons post hoc test; n = 3 biological replicates. (B) Postulated mechanism of action of CA in preventing Spike protein of SARS-CoV-2 from binding to ACE2. The reaction mechanism involves nucleophilic attack of cysteine thiol of ACE2 on the electron deficient carbon of the quinone formed by oxidation of CA.
reference link : https://www.mdpi.com/2076-3921/11/1/124/htm



{kind=link}