








")


 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Hanno pubblicato il loro articolo nel numero di marzo del Journal of Biological Chemistry.
Le infezioni gengivali gravi danneggiano i tessuti molli della bocca come le gengive ed erodono gradualmente le ossa (alveolari) sottostanti che sostengono i nostri denti. Sia le tasche ossee attorno alla base dei denti che i legamenti che ancorano i denti all’osso mascellare sono suscettibili di rompersi a causa di un’infezione batterica. Questa erosione ossea parodontale, non controllata, può alla fine causare la perdita dei denti.
I lipopolisaccaridi supportano la cellula batterica e proteggono dall’attacco delle cellule immunitarie, ma sono stati anche implicati nel causare l’infiammazione delle gengive attivando i recettori toll-like (TLR4) sulle cellule immunitarie che poi riconoscono i batteri come agenti patogeni.
Ad esempio, le cellule immunitarie come i neutrofili accumulati nei tessuti infiammatori potrebbero rilasciare dsRNA in bocca. Il recente studio ha studiato il dsRNA come sospetto nella progressione dell’infiammazione ossea durante la malattia parodontale.
Nelle ossa sane, gli osteoblasti stromali sulla superficie esterna di un osso depositano nuovo materiale osseo, mentre gli osteoclasti originati da cellule ematopoietiche distruggono il vecchio osso per il riassorbimento dei minerali; l’equilibrio tra le loro attività sostiene la massa ossea. Una proteina chiamata RANKL svolge un ruolo nel mantenimento di tale equilibrio e, quindi, nel modo in cui l’osso viene rimodellato con successo.
Utilizzando osteoblasti e cellule del midollo osseo di topi, oltre a una molecola sintetica analoga al dsRNA, gli autori dello studio hanno sperimentato l’esposizione delle cellule al dsRNA. Hanno osservato che il dsRNA ha chiaramente indotto la differenziazione di più osteoclasti, le cellule che distruggono l’osso.
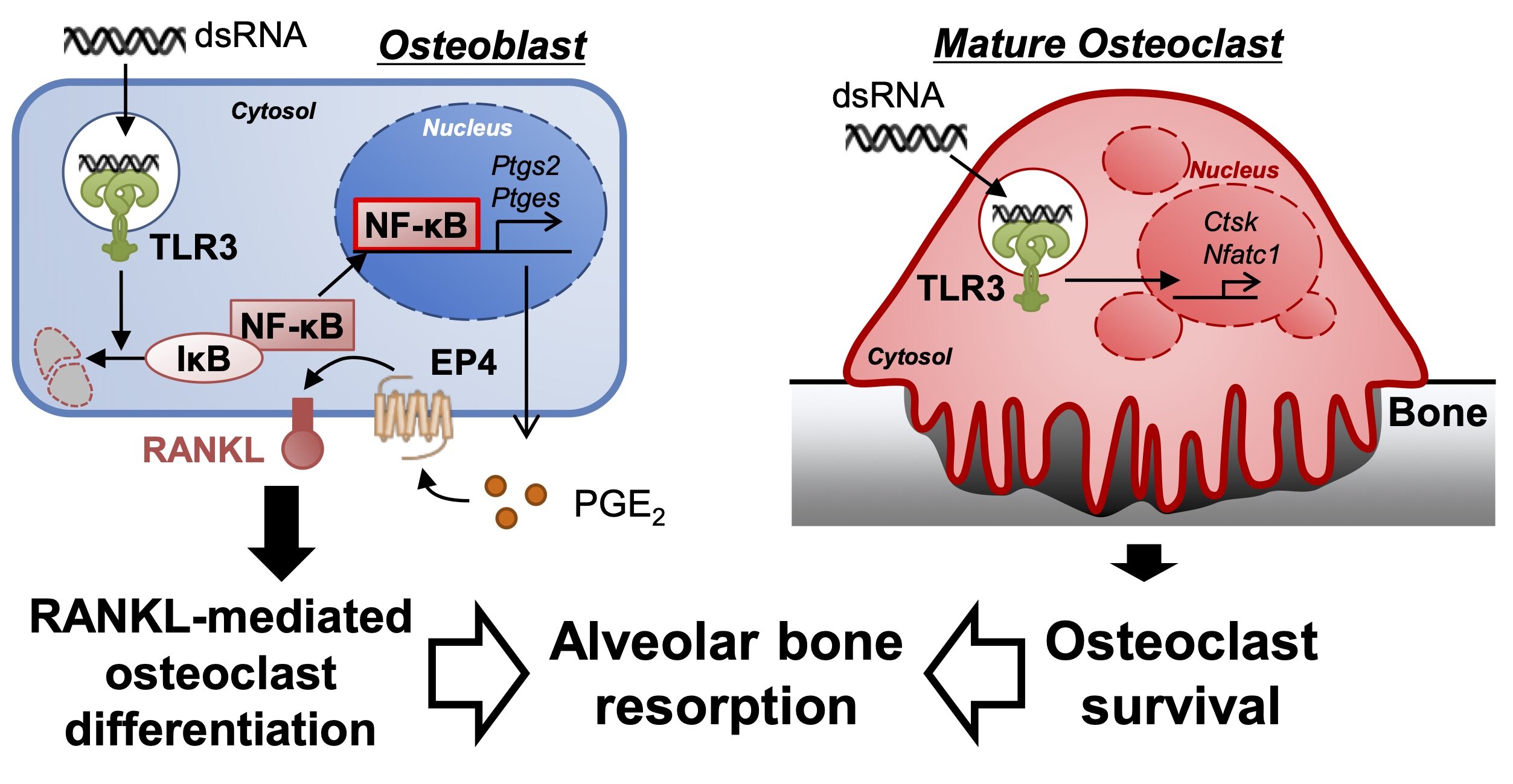
Il dsRNA ha indotto gli osteoblasti a produrre più PGE2 simile a un ormone che a sua volta ha sovraregolato RANKL e stimolato la differenziazione degli osteoclasti. Quindi, gli osteoblasti, attraverso le interazioni con le molecole di dsRNA, hanno inviato segnali cellulari che hanno aumentato la produzione degli osteoclasti che erodono l’osso. Il dsRNA ha anche fatto sopravvivere più a lungo gli osteoclasti maturi.
Più osteoclasti che sopravvivono più a lungo portano a un maggiore assorbimento dell’osso quando le gengive sono infiammate da malattie batteriche. Lo studio ha rivelato un meccanismo precedentemente sconosciuto mediante il quale la malattia gengivale provoca la rottura delle ossa. Dice Inada, “Questi dati suggeriscono che la segnalazione di TLR3 negli osteoblasti stromali controlla la produzione di PGE2 e induce la successiva differenziazione e sopravvivenza degli osteoclasti maturi”. Gli osteoclasti stromali portano al riassorbimento infiammatorio delle ossa che ancorano i denti. Sapere che l’infiammazione che porta al danno osseo nella parodontite può essere scatenata dal dsRNA introdotto tramite i batteri o dall’accumulo di cellule immunitarie nei tessuti è un balzo in avanti nella lotta agli effetti delle malattie gengivali.
Guardando al futuro, i ricercatori hanno in programma di esaminare ulteriormente come il dsRNA, segnalando ai recettori del sistema immunitario sugli osteoblasti stromali di produrre più PGE2, contribuisce alla progressione della parodontite nel tempo.
La malattia parodontale è una malattia infiammatoria locale associata a un’infezione batterica che provoca il riassorbimento dell’osso alveolare e la perdita dei denti. L’accumulo di placca batterica nella tasca parodontale porta alla patogenesi e alla progressione della malattia parodontale.
Il lipopolisaccaride (LPS), un componente della membrana dei batteri gram-negativi, è un noto pattern molecolare associato ai microbi che causa la malattia parodontale infiammatoria (1). Nel nostro studio precedente, che riportava un modello sperimentale di malattia parodontale, la produzione di prostaglandine (PG) E2 mediava una grave perdita ossea (2).
I topi privi di PGE sintasi legata alla membrana (mPGES)-1, un enzima inducibile per la sintesi di PGE, non sono riusciti a sviluppare la perdita di osso alveolare mediante iniezione di LPS, suggerendo che la sintesi di PGE2 indotta da mPGES-1 è essenziale per lo sviluppo mediato da LPS della malattia parodontale .
L’equilibrio tra riassorbimento osseo osteoclastico e formazione ossea osteoblastica mantiene la massa ossea. Per la differenziazione degli osteoclasti, l’attivatore del recettore del ligando NF-κB (RANK) (RANKL), che appartiene alla superfamiglia del fattore di necrosi tumorale, è una molecola essenziale.
L’espressione di RANKL è indotta da diversi fattori, comprese le citochine infiammatorie sulla membrana degli osteoblasti (3, 4, 5). I macrofagi ematopoietici che esprimono RANK come cellule precursori degli osteoclasti interagiscono con gli osteoblasti stromali che esprimono RANKL. L’interazione RANK-RANKL attiva diverse vie di segnalazione, come la via MAPK/AP-1, la via NF-κB e la via del calcio, con conseguente attivazione del fattore nucleare delle cellule T attivate, citoplasmatico 1 (NFATc1), un fattore di trascrizione principale per la differenziazione degli osteoclasti.
NFATc1 induce quindi varie proteine marcatori degli osteoclasti, come fattori di fusione cellulare tra cui la proteina transmembrana stimolante degli osteoclasti e la proteina transmembrana specifica delle cellule dendritiche, nonché proteasi, catepsina K e fosfatasi acida tartrato-resistente (TRAP).
Le cellule precursori degli osteoclasti poi si fondono tra loro e si differenziano in osteoclasti che riassorbono l’osso (6, 7, 8).
PGE2 è considerato un importante mediatore infiammatorio attraverso la sovraregolazione di RANKL nella malattia parodontale (2). La risposta infiammatoria sovraregola la sintesi degli enzimi associati alla produzione di PGE2, vale a dire, fosfolipasi citosolica A2, ciclossigenasi (COX)-2 e mPGES-1.
Gli acidi arachidonico vengono rilasciati dalla membrana fosfolipidica dalla fosfolipasi citosolica A2, quindi COX-2 converte l’acido arachidonico in PGH2 e mPGES-1 media la sintesi di PGE2 da PGH2. I recettori EP (EP1, EP2, EP3 e EP4) sono identificati come recettori PGE2.
Abbiamo riportato che EP2 ed EP4 sono coinvolti nel riassorbimento osseo mediato da PGE2 ed EP4 è segnalato come il principale recettore associato al riassorbimento osseo mediato da PGE2 (9, 10, 11). Studi precedenti hanno dimostrato che le molecole infiammatorie, come LPS e interleuchine infiammatorie, hanno aumentato la produzione di PGE2 attraverso il percorso NF-κB.
PGE2 legato a EP4 migliora quindi l’espressione di RANKL attraverso l’attivazione del fattore di trascrizione CREB negli osteoblasti, portando alla differenziazione degli osteoclasti (10, 11, 12, 13, 14).
I recettori toll-like (TLR) svolgono un ruolo essenziale nella risposta immunitaria innata, riconoscendo vari componenti microbici (15). Nei mammiferi, TLR1, 2, 4, 5 e 6 sono espressi sulla superficie cellulare, mentre TLR3, 7, 8 e 9 si localizzano sulla membrana endosomiale. LPS, un componente della membrana esterna dei batteri gram-negativi, è un potente induttore della malattia parodontale.
La segnalazione LPS e TLR4 promuove la produzione di citochine proinfiammatorie nei tessuti parodontali (16), stimola l’espressione di RANKL mediata da PGE2 negli osteoblasti stromali e inibisce la differenziazione osteogenica nelle cellule staminali del legamento parodontale (17), portando al riassorbimento infiammatorio dell’osso alveolare.
È stato dimostrato che ligandi sintetici per TLR2/1 e TLR2/6, Pam3CSK4 e Pam2CSK4 inducono la differenziazione degli osteoclasti mediata da PGE2 e la distruzione dell’osso alveolare in un modello murino di malattia parodontale (18). L’espressione di TLR1 è riportata anche nei tessuti parodontali (19, 20) ed è stato riportato che TLR5, che ha riconosciuto la flagellina batterica, induce la differenziazione degli osteoclasti e la perdita ossea (21).
Questi rapporti indicano che i TLR situati sulla superficie cellulare sono coinvolti nella perdita infiammatoria dell’osso alveolare; tuttavia, c’è una piccola prova del ruolo dei TLR endosomiale nel riassorbimento osseo infiammatorio, come la malattia parodontale. Uno dei TLR endosomiale, il TLR3 nelle cellule immunitarie ematopoietiche, riconosce l’RNA a doppio filamento (dsRNA) derivato da virus, batteri e cellule morte (22, 23, 24, 25, 26).
La segnalazione di TLR3-dsRNA attiva la segnalazione di NF-κB e dell’interferone di tipo I, portando a risposte immunitarie innate. Recentemente, l’acido poliinosinico-poliinocitidilico [poly(I:C)] è stato ampiamente utilizzato come ligando TLR3 per lo studio della segnalazione di TLR3. Poly(I:C) ha promosso lo sviluppo dell’artrite inducendo la produzione di citochine proinfiammatorie tramite la segnalazione TLR3 (27, 28); tuttavia, non è chiaro se la segnalazione TLR3 svolga un ruolo nelle malattie infiammatorie, come il riassorbimento osseo parodontale nella parodontite.
Nel presente studio, abbiamo esaminato i ruoli di TLR3 e del ligando nel riassorbimento osseo infiammatorio. Poly (I: C) si è interiorizzato nell’endosoma e ha indotto l’espressione di RANKL mediata da PGE2 attraverso la via di segnalazione NF-κB negli osteoblasti stromali, portando alla differenziazione degli osteoclasti. Poly(I:C) ha anche prolungato la sopravvivenza degli osteoclasti maturi differenziati.
I nostri dati indicano che la segnalazione TLR3 mediata da poli(I:C) negli osteoblasti stromali e negli osteoclasti contribuisce in modo collaborativo allo sviluppo della perdita ossea alveolare nella malattia parodontale.
link di riferimento: https://www.jbc.org/article/S0021-9258(22)00043-6/fulltext
Ulteriori informazioni: Tsukasa Tominari et al, Endosomal TLR3 signaling negli osteoblasti stromali induce il riassorbimento osseo parodontale infiammatorio mediato dalla prostaglandina E2, Journal of Biological Chemistry (2022). DOI: 10.1016/j.jbc.2022.101603



{kind=link}