










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Coronaviruses infect humans by binding to specific proteins, known as receptors, on human cell surfaces.
Researchers from the University of Minnesota, led by Professor Fang Li in the Department of Veterinary and Biomedical Sciences at the College of Veterinary Medicine, recently broke new ground in understanding how SARS-CoV-2, the virus that causes COVID-19, binds to its human receptor.
Their findings were recently published in the journal Nature.
This knowledge not only facilitates a better understanding of the infectivity of the virus, but also sheds light on its animal origin and provides guidance on vaccine and antiviral drug designs.
Several technologies, all with intrinsic limitations, have recently provided some information on how SARS-CoV-2 binds to its receptor.
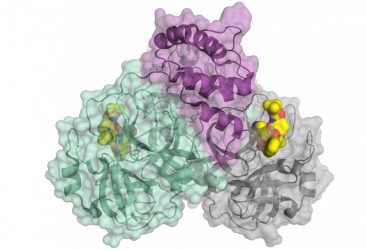
Li’s study is the first to use x-ray crystallography, the “gold-standard” method of structural biology at atomic resolution, to map out the 3D structure of a protein on SARS-CoV-2 that binds to its human receptor.
This 3D imagery reveals much information about how the two proteins connect.
More specifically, the 3D structure shows that:
- compared to the virus that caused the 2002-2003 SARS outbreak, the new coronavirus has evolved new strategies to bind to its human receptor, resulting in tighter binding of the receptor; and
- two related coronaviruses, one found in bats and one found in pangolins, can bind directly to the same human receptor as SARS-CoV-2, suggesting that SARS-CoV-2 originates from bats, either directly or with pangolins as an intermediate host. To infect humans, however, the bat or pangolin coronavirus needed to undergo mutations to gain more efficient usage of the human receptor.
The study has identified key mutations that potentially enabled the animal-to-human transmission of SARS-CoV-2.
With the 3D structure in hand, the study has mapped out the important binding sites on SARS-CoV-2 for antibody drugs to act on, providing a blueprint for developing new antibody drugs that specifically target those sites.
If a new antibody drug can bind to those sites on the virus more strongly and frequently than the receptor, it will block the virus out of cells, making it a potentially effective treatment for viral infections.
Those sites are also valuable for vaccine designs, as vaccines containing those sites can induce the production of antibodies in the human body, which can prevent future viral infections.
Several technologies, all with intrinsic limitations, have recently provided some information on how SARS-CoV-2 binds to its receptor.
Next, the research team plans to use structural information from this study to develop antibody drugs and vaccines that specifically target the binding of SARS-CoV-2 to its human receptor.
Other contributors to the study are Jian Shang, Gang Ye, Yushun Wan, Chuming Luo, Qibin Geng, Ashley Auerbach from Department of Veterinary and Biomedical Sciences, and Ke Shi, Hideki Aihara from Department of Biochemistry, Molecular Biology and Biophysics in the College of Biological Sciences and the Medical School. The study was funded by the National Institute of Health.
As part of continued efforts to respond to the COVID-19 pandemic caused by SARS-CoV-2 virus, researchers have used X-ray crystallography to reveal the structure of the SARS-CoV-2 main protease (SARS-CoV-2Mpro) – one of the best characterised drug targets among coronaviruses.
They used this information to identify a compound that binds to and inhibits the protease in mice.
The scientists, from Germany, say their study could drive the improved design of inhibitors to combat the novel coronavirus, a step toward urgently needed therapies to fight the global pandemic.
They focused on this protease due to its role in processing polyproteins that are translated from viral RNA.
Based on studying the structure of the main protease, the researchers optimised inhibitors for existing coronaviruses to develop compound 13b, a potent blocker of the SARS-CoV-2 main protease.
Using the X-ray structures of SARS-CoV-2 without the ligand, they studied its complex with an α-ketoamide inhibitor derived from a previously designed compound but with the P3-P2 amide bond incorporated into a pyridone ring to enhance the half-life of the molecule in blood.
Lead researcher Linlin Zhang and colleagues found that a high-resolution structure of 13b bound to the protease and blocked viral RNA replication. They report that 13b has features that improve over existing inhibitors, including an extended half-life in blood plasma.
They further tested their leading inhibitor compound in mice, finding that inhalation was well tolerated and that the mice did not show any adverse effects.
The researchers suggest that as no human proteases with a similar cleavage specificity are known, this class of inhibitors are unlikely to be toxic.
The team suggest that their results show direct administration of the compound to the lungs may be possible and provide a framework for the development of drugs to combat the novel coronavirus.
“Given these favourable pharmacokinetic results, our study provides a useful framework for development of the pyridone-containing inhibitors toward anticoronaviral drugs,” the authors conclude.

Source:
University of Minnesota



{kind=link}
{kind=link}
[…] COVID-19 has evolved new strategies to bind to cell receptors –… […]
[…] COVID-19 has evolved new strategies to bind to cell receptors –… […]