









 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



For decades, biology textbooks have stated that eyes communicate with the brain exclusively through one type of signaling pathway. But a new discovery shows that some retinal neurons take a road less traveled.
New research, led by Northwestern University, has found that a subset of retinal neurons sends inhibitory signals to the brain. Before, researchers believed the eye only sends excitatory signals. (Simply put: Excitatory signaling makes neurons to fire more; inhibitory signaling makes neurons to fire less.)
The Northwestern researchers also found that this subset of retinal neurons is involved in subconscious behaviors, such as synchronization of circadian rhythms to light/dark cycles and pupil constriction to intense bright lights.
By better understanding how these neurons function, researchers can explore new pathways by which light influences our behavior.
“These inhibitory signals prevent our circadian clock from resetting to dim light and prevent pupil constriction in low light, both of which are adaptive for proper vision and daily function,” said Northwestern’s Tiffany Schmidt, who led the research.
“We think that our results provide a mechanism for understanding why our eye is so exquisitely sensitive to light, but our subconscious behaviors are comparatively insensitive to light.”
The research will be published in the May 1 issue of the journal Science.
Schmidt is an assistant professor of neurobiology at Northwestern’s Weinberg College of Arts and Sciences. Takuma Sonoda, a former Ph.D. student in the Northwestern University Interdepartmental Neuroscience program, is the paper’s first author.
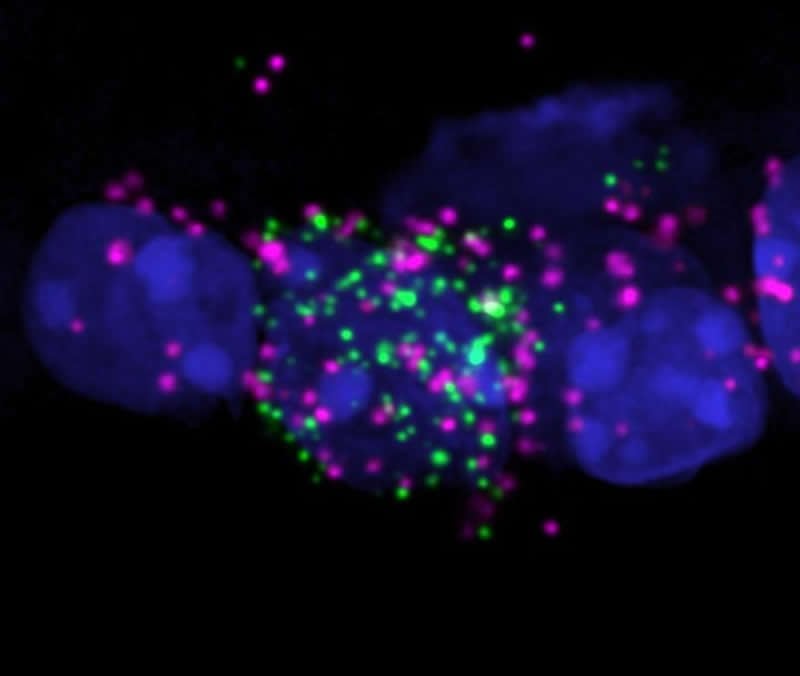
To conduct the study, Schmidt and her team blocked the retinal neurons responsible for inhibitory signaling in a mouse model. When this signal was blocked, dim light was more effective at shifting the mice’s circadian rhythms.
“This suggests that there is a signal from the eye that actively inhibits circadian rhythms realignment when environmental light changes, which was unexpected,” Schmidt said.
“This makes some sense, however, because you do not want to adjust your body’s entire clock for minor perturbations in the environmental light/dark cycle, you only want this massive adjustment to take place if the change in lighting is robust.”
Schmidt’s team also found that, when the inhibitory signals from the eye were blocked, mice’s pupils were much more sensitive to light.
“Our working hypothesis is that this mechanism keeps pupils from constricting in very low light,” Sonoda said. “This increases the amount of light hitting your retina, and makes it easier to see in low light conditions. This mechanism explains, in least part, why your pupils avoid constricting until bright light intensifies.”
Funding: The research, “A non-canonical inhibitory circuit dampens behavioral sensitivity to light,” was supported by a Klingenstein-Simons Fellowship in the Neurosciences, the Alfred P. Sloan Foundation and the National Institutes of Health (award numbers 1DP2EY022584, T32 EY025202 and F31 EY030360-01).
Introduction to the molecular circadian clock
Circadian rhythms evolved as organisms were selected for their preparedness for the environmental shifts brought about by the solar day (8). The molecular model for circadian rhythm generation arose from genetic screens designed to find mutations that disturb the sleep-wake cycle in model organisms (7).
In both flies and mammals, the key constituents of the molecular circadian clock are either transcription factors or transcription factor regulators (i.e., “clock genes”; Figure 2). At the core of the clock is a heterodimer composed of the proteins BMAL1 and CLOCK (9).
This complex stimulates transcription by altering the chromatin landscape around genes containing E-box motifs in their promoters (10). Among the downstream effectors of BMAL1/CLOCK are fellow clock gene products Per1–3, Cry1/2, NR1D1/2 (also known as REV-ERBα/β, and RORα–γ, whose products either negatively or positively regulate the core complex (9).
Thus, the expression of clock genes oscillates with roughly a 24-hour periodicity, thereby biochemically representing the solar day. Other downstream effectors of the molecular clock include master transcription factors, such as the PAR domain basic leucine zipper transcription factor family (DBP, TEF, and HLF) (11), nuclear factor IL-3–regulated (NFIL3, also known as E4BP4) (12), and the PPAR family (11, 13).
Through these clock-controlled genes (CCGs) as well as others, the molecular clock imparts a circadian pattern on gene expression, and by extension protein and metabolite abundance. Molecular clocks operate in almost all nucleated cells throughout the body, thereby allowing cells to factor time-of-day information into the control of metabolism and other key pathways (14).

Object name is jciinsight-5-131487-g200.jpg
Figure 2
Circadian regulation in mammals.
Schematic depicting the currently accepted hierarchal model for circadian rhythm generation. Light information is conveyed by the optic nerve to the SCN, a region of the ventral hypothalamus. There, light entrains clocks within SCN neurons, and this is ultimately converted by the CNS into pulsatile chemical and neurological cues, which entrain cell-autonomous circadian clocks residing in peripheral cells. These peripheral clocks impart circadian patterns on gene expression and overall cellular physiology. For simplicity, only the core molecular clock circuitry is depicted, with positive regulatory proteins labeled green and negative regulators labeled red. However, there are many accessory proteins and metabolic pathways that can adjust the periodicity and phase of the clock but are not central to rhythm generation (for example, casein kinase 1δ/ε [ref. 23], AMPK [ref. 149], mTOR [ref. 150], p53 [ref. 151], and SIRT1 [ref. 152]). There are additional molecular clock constituents (not depicted) that in the basal state appear to have more prominent roles in CNS clocks than clocks in peripheral cells. For example, NPAS2 (a functional homolog of CLOCK) and DEC1/2 provide additional negative feedback to BMAL1/CLOCK (16, 153). Yellow boxes represent E-boxes or ROR-responsive elements (RREs), which are the promoter motifs recognized by BMAL1/CLOCK or REV-ERB/ROR proteins, respectively. Clock genes and clock-controlled genes (CGs) are represented by green and purple arrows, respectively. Illustrated by Rachel Davidowitz.
The molecular clock contains some special features that are important for its role in regulating the physiology of higher organisms. First, clock proteins and clock genes are subject to numerous forms of biochemical regulation, including acetylation, methylation, phosphorylation, ubiquitination, and sumoylation (15).
Combined with signal transduction through accessory pathways, these modifications impact clock protein levels and activity, thereby allowing ambient biochemical cues, such as temperature, nutrient status, hormone levels, and innervation, to synchronize internal cellular time with the external world (16).
This phenomenon is known as entrainment and serves an additional crucial role in creating temporal coherence within organs by synchronizing parenchymal cells with one another. To harmonize the various biochemical entrainment cues, mammals require a “master circadian pacemaker” located in the suprachiasmatic nucleus (SCN) of the hypothalamus.
The SCN receives light information from the retina and then projects to other CNS areas that regulate arousal, autonomic tone, temperature, and hormone secretion (16). Of all the entrainment cues, light has the largest impact on rest-activity rhythms, and the clocks within SCN neurons are selectively sensitive to light for entrainment.
However, because SCN clocks synchronize to changing light cycles faster than peripheral clocks, and because mistimed feeding or other biochemical cues can compete with SCN pacing in the periphery (17), it is possible to induce a state of internal desynchronization.
This desynchronization is thought to explain the sequelae of jet lag and night-shift work (18–20). A second key feature of the molecular clock is that its regulatory influence extends from the microscopic to the macroscopic biological scale.
For example, mutations that alter the periodicity of Per and Cry expression provoke analogous changes to the periodicity of sleep timing and quality (21–23). A final feature is programmability. While the anatomy of the molecular clock is largely uniform across tissues, the circadian transcriptomes it generates (i.e., the set of CCGs exhibiting rhythmic expression) are highly tissue specific and overlap by only about 10% between organs (24).
The remarkable organ specificity of circadian transcriptomes is thought to be a product of cell type–specific transcription factors and tissue-selective patterns of chromatin accessibility (25, 26).
Collectively, the cardinal molecular clock features of entrainment, programmability, and the ability to impact function at all biological scales enable tissues to harness time-of-day information in the service of their unique physiologic contributions. They also makes it possible for the clock to organize diverse networks of cells around a common goal, for example, a coordinated immune response.
Clocks and immunity
The current focus on circadian control of immune function was presaged by the observations — first published more than 50 years ago — that circulating lymphocyte counts oscillate on a daily basis in healthy humans (27) and that the susceptibility to endotoxin depends on the time of day it is administered in rodents (28).
More recent work highlights epidemiological associations between night-shift work (i.e., activity and sleep patterns in opposition to the normal day-night pattern) and elevated levels of systemic inflammatory markers (29, 30).
Significantly, all lymphoid organs and immune cell types tested to date harbor functional clocks (31, 32). Studies using genetic experimental approaches in human cells and mice suggest a connection between clocks and at least three fundamental immunologic activities.
The best-documented connection between clocks and immune function is the regulation of proinflammatory cytokine secretion.
Cytokines are well-known mediators of host responses to infection, trauma, and immune responses. In endotoxin-stimulated macrophages, the magnitude of TNF-α secretion was found to vary in a circadian fashion based on the time of endotoxin challenge (31).
The current paradigm for explaining how the clock imparts rhythms to cytokine production involves the ability of clock proteins to directly transactivate or repress gene expression of key cytokines and chemokines, including CCL2, TNF, IL-6, and CXCL5 (reviewed in ref. 33). This paradigm also extends to CCGs.
For example, in LPS-stimulated macrophages, the clock-controlled transcription factors NFIL3 and DBP impart a circadian rhythm to IL-12(p40) secretion by alternating their binding to the Il12 promoter throughout the day (34). Thus, disruption of molecular clock function through light-cycle desynchronization or deletion of Bmal1 or Reverba locks macrophages into a “time of day” wherein inflammatory cytokine secretion is maximized (35, 36).
This hypersecretory phenotype is not limited to immune cells but extends to epithelial cells, which are responsible for initial leukocyte recruitment after infectious and sterile insults (36–38). Interestingly, inflammation appears to feed back on the integrity of the molecular clock. For example, Clock, REV-ERBα, and BMAL1 protein levels decline rapidly in cells after endotoxin treatment, which is predicted to disrupt clock function (39–41).
Conversely, herpes virus infection stimulates Bmal1 promoter activity, leading to a corresponding depression in Cry1 and a disruption in the feedback relationships that underlie the clock (42). Indeed, disruption of clock gene expression appears to be a common outcome of acute infection and leads to novel rhythms of gene expression characterized by prominent cycling of proinflammatory genes (37, 43).
Some suggest that the circadian clock is itself a kind of innate immune sensor that when disabled by infection automatically shifts cells to a proinflammatory state. However, during chronic inflammation or stressors such as shift work, disrupted clocks have been shown to increase the morbidity in response to infection or sterile insult through excessive inflammation (44–49).
Trafficking of myeloid and lymphocyte subsets in healthy mice and humans is another circadian-regulated immune activity (50–52). Leukocyte trafficking represents the summation of several discrete, individually rhythmic processes (Figure 3), including cell egress from bone marrow (53, 54), leukocyte homing from the bloodstream to the interstitial space of organs (51, 55), and removal of leukocytes from organs through phagocytosis or via transit to regional lymph nodes (56, 57).
Regardless of cell type, mechanisms of rhythmic trafficking involve coordinated clock function within the leukocyte and within endothelial cells lining the vessels of peripheral organs. Together, these clocks align the expression of receptor-ligand pairs critical for chemoattraction and adhesion to the endothelial surface, thereby facilitating leukocyte recruitment (51, 55). A
dditional levels of control, including adrenergic tone and leukocyte ROS, appear to play a role in setting the phase of leukocyte trafficking rhythms (51, 52, 58). Interestingly, the critical ligand-receptor interactions needed for leukocyte homing appear to vary depending on the cell type and the destination organ (55). Altogether, the complexity of leukocyte trafficking makes clear the importance of the clock in optimizing this critical immune activity.

Multilayered circadian control of leukocyte trafficking.
Schematic depiction of the circadian regulation of specific leukocyte trafficking steps (55–57, 68). These include the egress of leukocytes from the bone marrow; adhesion of circulating leukocytes to endothelial cells in the capillary beds of end organs; and removal of leukocytes from the parenchyma of organs by phagocytosis or by migration to area lymph nodes. Proteins generally important for rhythmic leukocyte trafficking at specific steps in the process are depicted in red. For a comprehensive treatment of cell-specific determinants of leukocyte trafficking rhythms, see Pick et al. (154). Lymph/Mac, lymphocyte/macrophage. Illustrated by Rachel Davidowitz.
A third connection between clocks, clock genes, and immune processes is among the least studied: differentiation and maturation of clinically important leukocyte subsets. For example, the canonical clock gene Rora is required for the differentiation of type 2 innate lymphoid cells (ILC2s), which play a role in type II inflammation, allergy, and defense against parasites (59).
A splice variant of the clock gene Rorg (Rorgt) is critical for the development of type 3 innate lymphoid cells (ILC3s), lymphoid tissue inducer (LTi) cells, and the CD4+ T helper cell subset Th17 cells (60–62). Rora is also important for the full development of Th17 cells (63). ILC3s and Th17 cells are characterized by secretion of IL-17, and both are implicated in autoimmune diseases.
Disruption of molecular clock function leads to aberrant Th17 cell development and renders mice more susceptible to pathology in models of autoimmunity, including experimental autoimmune encephalomyelitis (EAE), a mouse multiple sclerosis model, and colitis (64, 65). Interestingly, Rorgt mRNA expression is blocked by melatonin, a hormone with pleiotropic immunomodulatory activity that is secreted in a circadian fashion by the pineal gland (66, 67).
Finally, circadian clocks were recently found to regulate neutrophil maturation (68), which is remarkable given the short life span (only 1–2 days) of these crucial effector cells. As neutrophils age, they undergo cytoskeletal and cell-adhesive changes that enhance their migration into the interstitial spaces of organs according to a circadian rhythm (53, 68).
These changes are mediated by Bmal1, and loss of this clock gene blocks their efficient homing to organs (68). The result is that initial innate responses against invading organisms are weakened in Bmal1-deficient mice, thereby increasing disease severity (68).
Clock control of neutrophil maturation has an additional homeostatic function, as trafficking of aged neutrophils into the bone marrow suppresses the hematopoietic niches that control the egress of leukocytes in the first place (53). In doing so, rhythmic neutrophil homing completes a feedback loop that may help to make circadian trafficking patterns for all hematopoietic types self-sustaining.
Altogether, it is increasingly clear that circadian gating is part of the core programing of the immune system, and thus, alteration of this regimen is likely to have widespread ramifications for disease pathogenesis. Current evidence draws a direct mechanistic line between the biochemistry of a clock protein, such as BMAL1, and a discrete immune parameter, like the production of the neutrophil chemoattractant CXCL5 during lung injury (36). Alternatively, the clock may impact immunity by regulating sleep.
Sleep and immunity
Sleep is a basic behavior common to almost all organisms and is defined by a period of relative unresponsiveness to outside stimuli. In humans, sleep can be subdivided into an orderly progression of neurological stages based on EEG patterns that repeat roughly every 90 minutes (69).
There is no consensus on what defines ideal sleep, but total sleep deprivation in rodents is fatal (70). It is still not fully understood why denying rodents sleep is deadly; however, studies have shown that chronically sleep-deprived animals exhibit splenic atrophy and polymicrobial bacteremia, suggesting that immune dysfunction may be part of the dying process (70, 71).
In a similar vein, short habitual sleep (<6 hours per night) is statistically associated in humans with reduced life span, increased vulnerability to viral infection, and reduced antibody titers after vaccination (72–74). Short-term sleep deprivation prior to vaccination appears to negatively impact antibody titers after influenza vaccination and, at least in rodents, reduces influenza vaccine efficacy (75–77).
Just as sleep impacts immune functions, it appears that certain peripheral immune populations may affect sleep. For example, chemical or genetic ablation of macrophages affects the architecture of rebound sleep in sleep-deprived mice (78, 79). Altogether, sleep is connected to an organism’s resilience against infection, and considerable work has been done in humans and animal models to examine how sleep deprivation impacts specific immune parameters (80).
The relationship between circadian rhythms and sleep is complex and likely bidirectional. In humans, the paradigm for sleep timing is a “two-process” model (processes C and S) in which CNS circadian clocks play a major role. Clocks generate daily oscillations in neurological arousal (process C), which promotes wakefulness and opposes drowsiness that accumulates through sleep debt (process S) (81).
Work in Bmal1-deficient model organisms suggests that clocks may have an additional role in mediating normal sleep architecture (82–84). While clocks gate sleep, it is also true that short-term sleep disruption can affect circadian regulation in peripheral tissues such as blood and lung, by either damping the rhythms in CCG expression or altering their phase (85, 86).
Thus, while circadian rhythms and sleep are biologically different phenomena and can be analyzed in isolation using “forced synchronization” protocols in humans (87), it is difficult to perturb one without affecting the other under naturalistic conditions.
For physicians, the conflation of circadian rhythms and sleep during disease dates to Greek antiquity. The writings of Arataeus of Cappadocia on asthma posed the question of whether asthma exacerbations are more frequent at night because of the time of day or because patients are typically asleep at night (88).
Some pathogens even corrupt the circadian-mediated sleep/wake cycle to promote disease. For example, Trypanosoma brucei (which causes African sleeping sickness) alters sleep patterns in afflicted subjects by shortening the periodicity of the molecular circadian clock (89).
Given the interconnectedness of sleep and circadian rhythms, it is understandable that the immune activities affected by insufficient sleep overlap with those altered by circadian disruption.
Experimentally induced sleep deprivation and short habitual sleep increase proinflammatory cytokine secretion, particularly in males (90, 91). Numbers of circulating neutrophils, NK cells, monocytes, and B cells are increased by prolonged wakefulness and decreased by recovery sleep, suggesting a role for sleep in regulating leukocyte trafficking (80, 92).
Recent work in mice found that sleep fragmentation increases the production of inflammatory monocytes by the bone marrow and leads to atherosclerosis, an inflammatory pathology also induced by Bmal1 deficiency (93, 94). Finally, inadequate sleep seems to impair immune effector cell functions such as NK cell activity, at least in some experimental scenarios (80, 95).
Circadian rhythms and sleep are increasingly seen as important for immune system homeostasis. Disruption of either process can engender a state of inflammation and functional immunocompromise, rendering organisms more vulnerable to disease.
Historically, circadian biology, sleep, and immunology have occupied separate branches of investigation. Consequently, there has been little interdisciplinary research on these topics.
The key point is that circadian rhythm and sleep disruption happen concurrently every day through the widespread experiences of shift work, nighttime light exposure, and social jet lag, which is caused by discontinuity between natural sleep-wake schedules and the variations in the social demands of weekdays versus weekends. Thus, the interface between circadian rhythms, sleep, and immune function potentially has a wide range of implications for public health and medicine.
Source:
Northwestern University



{kind=link}