











 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Every second of every day, countless biochemical reactions take place in our bodies’ cells.
The organization of this complex system is the result of billions of years of evolution, fine-tuning our functions since the first primordial organisms.
One such vital reaction is ‘methylation’, where a methyl group – a carbon atom linked to three hydrogen atoms – attaches itself to a target molecule. Methylation is involved in the regulation of everything from DNA to proteins, and it is so vital that it can be found in all living organisms.
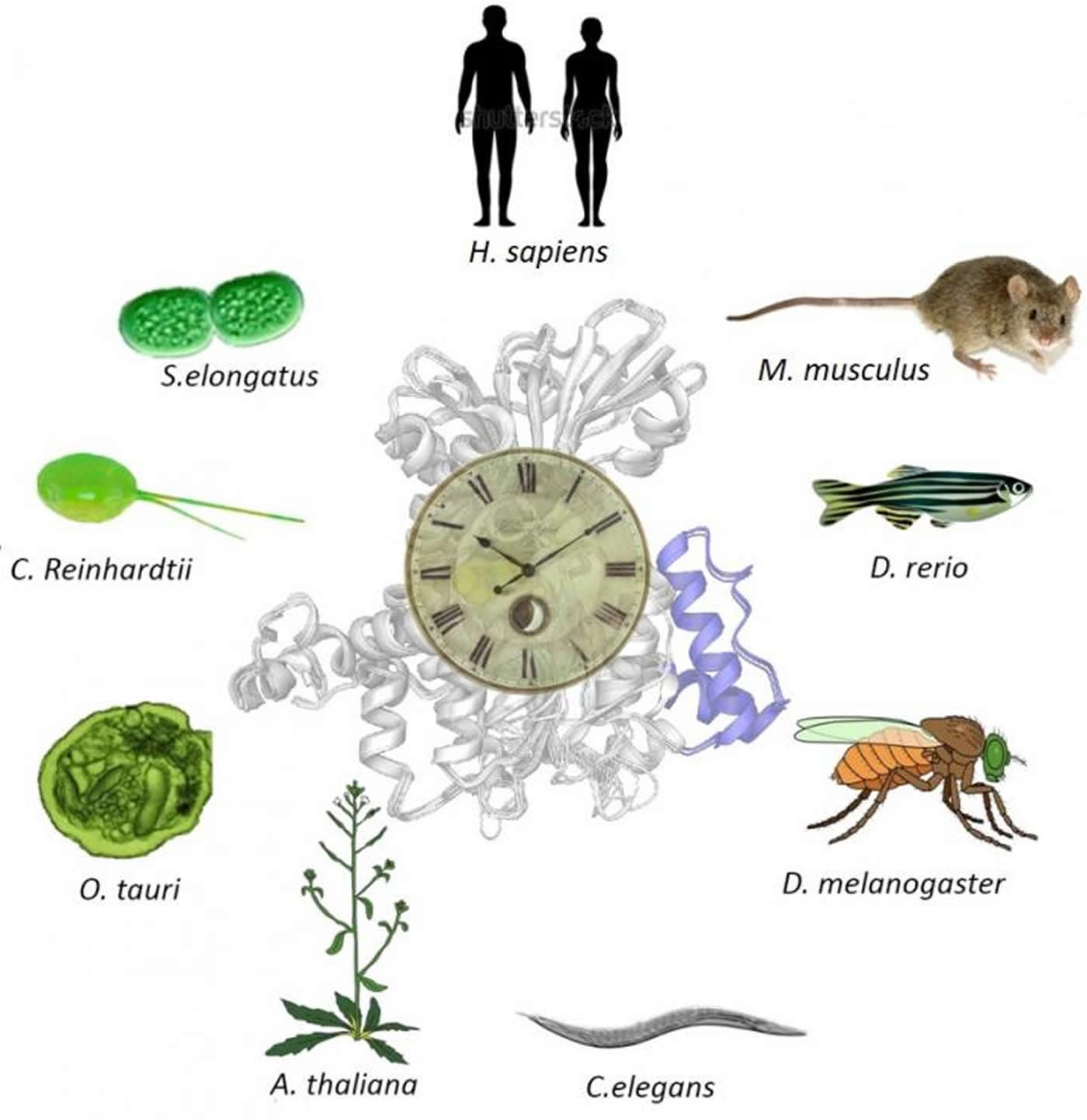
In a recent paper published in Communications Biology, a team of researchers lead by Jean-Michel Fustin and Hitoshi Okamura from Kyoto University’s Graduate School of Pharmaceutical Sciences has uncovered an intimate connection between methylation and the body’s circadian rhythms: a link that exists even in organisms that don’t traditionally ‘sleep’, such as bacteria.
“Disfunction in methylation can cause any number of pathologies, from atherosclerosis to cancer,” explains Fustin. “Previously we discovered that inhibiting methylation in mice and human cells disrupted their body clocks.”
Methylation and the circadian rhythm, he adds, are ancient mechanisms retained in many organisms from bacteria to humans. “So, we hypothesized that the link between the two was also ancient.”
The team began by collecting cells and tissue samples from different organisms and measuring their biological rhythms. On average, all organisms run on periods of 24 hours.
The next step was to find out what happens when methylation is disrupted, and as anticipated, significant alterations in the circadian clock were detected in all cell types, including in plants and algae.
However, cyanobacteria – photosynthetic bacteria – seemed relatively resistant.
“The methylation pathway in bacteria is slightly different from other organisms. But when an alternative compound inhibiting a different part of methylation was used, the circadian clock was indeed strongly affected there as well,” Fustin continues.
Applying their findings, the team then took a gene that is key in controlling bacterial methylation and introduced it into mouse and human cells.
Exceptionally, the bacterial gene was able to protect the cells from the first methylation inhibition compound, with no alterations observed in circadian rhythms.
“Not only did we find the evolutionarily conserved link between two ancient biological pathways – methyl metabolism and biological clocks – but we also opened the door to a possible new treatment for methylation deficiencies,” concludes Okamura.
“All organisms are more alike than you might think, and knowledge about how we evolved will allow us to better understand ourselves and the natural world.”
Jean-Michel Fustin is currently affiliated with the University of Manchester.
Background
Methylation of mitochondrial tRNAs (mt-tRNA) at the 9th position (“p9 site”) is known to impact translational efficiency and downstream mitochondrial function; however, direct assessment of mt-RNA methylation is challenging. Recent RNA sequence-based methods have been developed to reliably identify post-transcriptional methylation. Though p9 methylation has been studied in healthy human populations and in the context of cancer, it has not yet been analyzed in neurodegenerative disease, where mitochondrial dysfunction is a prominent and early hallmark of disease progression.
Methods
Mitochondrial p9 methylation was inferred from multi-allelic calls in RNA-seq data. Gene-based association studies were performed in FUMA. Correlations between nuclear gene expression and p9 methylation were tested using Spearman’s rho. Fisher’s Exact test was used in PANTHER and IPA to test for overrepresentation and enrichment of biological processes and pathways in the top nuclear genes correlated with p9 methylation.
Results
Variable methylation was observed at 11 p9 sites in post-mortem cerebellar tissue of elderly subjects who were either healthy or diagnosed with Alzheimer’s disease (AD), progressive supranuclear palsy (PSP) or pathological aging (PA).
Similarities in degree of methylation were observed between AD and PSP. Certain nuclear encoded genes were identified as significantly associated with p9 methylation. Expression of 5300 nuclear encoded genes was significantly correlated with p9 methylation, with AD and PSP subjects exhibiting similar expression profiles. Overrepresentation and enrichment testing using the top transcripts revealed enrichment for a number of molecular processes, terms and pathways including many of which that were mitochondrial-related.
Conclusion
With mitochondrial dysfunction being an established hallmark of neurodegenerative disease pathophysiology, this work sheds light on the potential molecular underpinnings of this dysfunction. Here we show overlap in cerebellar pathophysiology between common tauopathies such as Alzheimer’s disease and progressive supranuclear palsy. Whether p9 hypermethylation is a cause or consequence of pathology remains an area of focus.
Conclusion
This is the first work analyzing mitochondrial p9 methylation in the context of neurodegeneration. Here we report an association between p9 methylation and nuclear-encoded gene expression, as well as mitochondrial-related regulation; although, the implications of these associations are unclear.
We observed comparable post-transcriptional mt-tRNA hypermethylation and nuclear gene expression profiles in the cerebellum of Alzheimer’s disease and progressive supranuclear palsy patients.
Although technical limitations in this analysis do not allow us to conclude on methylation status of the mature tRNA pool, the results here are likely indicative of molecular similarities underlying the mitochondrial dysfunction already known to occur within the cerebellum of individuals diagnosed with either of these tauopathies.
A basal level of p9 methylation is required for proper tRNA folding and stability [Helm M, Giegé R, Florentz C. A Watson−Crick Base-pair-disrupting methyl group (m1A9) is sufficient for cloverleaf folding of human mitochondrial tRNALys. Biochemistry. 1999;38(40):13338–13346.] and the degree of methylation at p9 sites has been shown by other groups to have low inter-individual variability [Ali AT, Idaghdour Y, Hodgkinson A. Nuclear genetic regulation of human mitochondrial RNA modification. bioRxiv. 2019;666339. 10.1101/666339.]. In addition, p9 hypermethylation has been observed in other age-related diseases such as cancer [Idaghdour Y, Hodgkinson A. Integrated genomic analysis of mitochondrial RNA processing in human cancers. Genome Med. 2017;9(1):36.].
We propose that p9 hypermethylation in AD and PSP may be more closely associated with pathology as opposed to normal aging. As shown in Fig. 6, we observed that specific nuclear encoded variants associated with hypermethylation at mitochondrial p9 sites within cerebellar neurons, importantly causality is not clear (i.e. associations may be linked to pathological processes and not necessarily RNA methylation).
For example, hypermethylation of p9 may be the primary event that impacts the function of the electron transport chain (ETC) and results in mitochondrial dysfunction, due to the influence that p9 methylation has on downstream mitochondrial protein translation.
Alternatively, it is possible that ETC dysfunction is the primary event that causes altered p9 methylation, in which case, the p9 hypermethylation is serving as an endophenotype of genetic risk for mitochondrial dysfunction. Regardless, through retrograde signaling, mitochondrial dysfunction may impact nuclear gene expression resulting in alterations in cellular and molecular processes to further exacerbate neurodegenerative pathology.

Hypothetical schematic of potential causes and effects of p9 methylation in neurodegeneration. (1) Nuclear encoded genetic variants may be causal for p9 hypermethylation within neuronal mitochondria, resulting in onset or exacerbation of electron transport chain dysfunction. (2) Subsequent mitochondrial dysfunction may lead to altered downstream nuclear gene expression via retrograde signaling resulting in (3) changes to various molecular and cellular processes that further contribute to neurodegenerative pathology
Though at present, it is unclear whether mt-tRNA methylation is a cause or consequence of pathology, these results point to a potential role for mitochondrial post-transcriptional methylation in the pathophysiology of common tauopathies such as Alzheimer’s disease and progressive supranuclear palsy. Given this, post-transcriptional mt-tRNA methylation may serve as an exciting area of investigation for the development of future treatment strategies for neurodegenerative disease.
Source:
Kyoto University



{kind=link}