










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



City of Hope scientists have combined two potent immunotherapies – an oncolytic virus and chimeric antigen receptor (CAR) T cell therapy – to target and eradicate solid tumors that are otherwise difficult to treat with CAR T therapy alone, according to a new Science Translational Medicine study.
In preclinical research that could lead to a clinical trial for patients with intractable solid tumors, City of Hope scientists genetically engineered an oncolytic virus to enter tumor cells and force their expression of CD19 protein on their cell surface.
Scientists were then able to use CD19-directed CAR T cells to recognize and attack these solid tumors.
CD19-CAR T cell therapy is approved by the U.S. Food and Drug Administration to treat certain types of blood cancers, namely B cell lymphomas and acute lymphoblastic leukemia.
This new research may expand the use of CD19-CAR T cells for the treatment of patients with potentially any solid tumor.
“Our research demonstrates that oncolytic viruses are a powerful and promising approach that can be combined strategically with CAR T cell therapy to more effectively target solid tumors” said Saul Priceman, Ph.D., the study’s senior author and an assistant professor in City of Hope’s Department of Hematology & Hematopoietic Cell Transplantation.
“In addition, this therapeutic platform addresses two major challenges that make solid tumors so difficult to treat with immunotherapy. There are limited, established solid tumor targets that T cells can be redirected against with CARs,” Priceman added.
“Furthermore, solid tumors are surrounded by a brick wall – a so-called immunosuppressive tumor microenvironment. When a CAR T cell attempts to enter the tumor, survive, and kill cancer cells, it can’t effectively because of this barrier.”
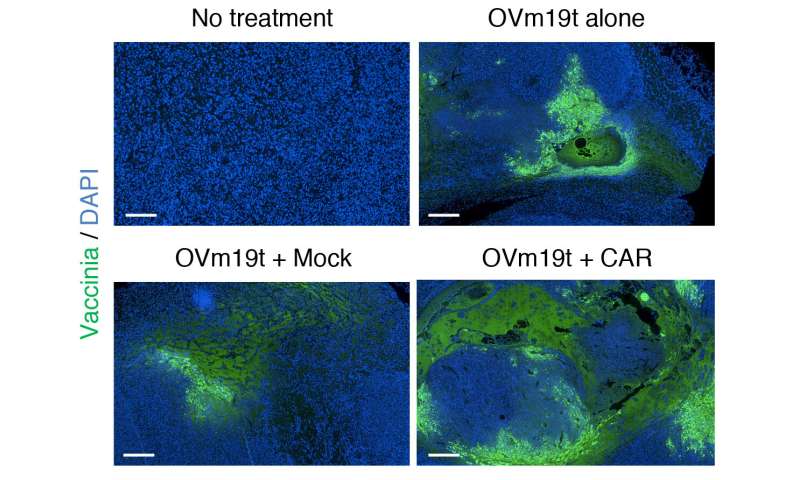
Yuman Fong, M.D., the Sangiacomo Family Chair in Surgical Oncology at City of Hope and a leading scientist who is developing oncolytic viruses for cancer treatment, added that the virus was able to break through that barrier.
“We designed this oncolytic virus to do what it does so well,” Fong said. “It entered the cancer cell and used the cell’s own machinery to replicate itself, and engineer the cancer cells to express a truncated form of the well-known CAR T cell target, CD19.”
Researchers first created an oncolytic virus (OV19t) in Fong’s lab to get into tumor cells and start producing truncated CD19 (CD19t). They did this successfully in triple-negative breast cancer lines, as well as in pancreatic, prostate, ovarian, and head and neck cancer, as well as brain tumor cells. CD19-CAR T cells were then combined with OV19t in vitro and in therapeutic studies in mice.
Researchers found several key findings.
“When we infected tumor cells with the virus, we observed the first signal that this may work. CD19t was being expressed by tumor cells much sooner than the virus was able to kill them, giving us a window of opportunity to be targeted by CD19-CAR T cells,” said Anthony Park, Ph.D., a postdoctoral fellow in Priceman’s lab. “The combination of the two had a powerful, synergistic effect.”
Researchers also showed that mice already cured of their cancer with the oncolytic virus and CAR T cell combination demonstrated prolonged protective anti-tumor immunity.
“The immune system built a memory response to the tumor,” Park added. “Once it eradicated tumors, following the initial combination treatment, the mice were shielded against tumor recurrences.”
Solid tumors are often immunologically cold, which means they are not typically responsive to therapies that use the body’s own immune system to fight cancer, Park said. Introducing the virus reversed the tumor’s harsh microenvironment, making it more receptive to receiving CAR T cell therapy.
The research demonstrates City of Hope’s collaborative approach to finding better immunotherapy cancer treatments. A few years ago, Priceman, Fong and Stephen Forman, M.D., leader of City of Hope’s Hematologic Malignancies Research Institute, met to brainstorm how they might combine their expertise, namely oncolytic virus and CAR T cell therapies, to target solid tumors.
“It was a simple concept but one that took many steps to get us to where we are today—we are now designing a clinical trial to test this combination in patients,” said Priceman.
The trial would first test the safety of OV19t in patients with solid tumors. If that is found to be safe and effective, the oncolytic virus and CAR T cell therapy could then be tested in sequence. The trial is anticipated to start in 2022.
Between 2003 and 2007, in 21 countries representative of four regions of the globe, the incidence rate of acute lymphoblastic leukemia (ALL) ranged from 2.17–3.78/100,000 individuals per year, in children younger than 19 years (1).
ALL is the most common form of childhood cancer, with a peak in incidence between 2 and 5 years of age (2,3). The revision of and improvements in drug combinations and the intensity of radiotherapy has improved the outcome of ALL. The 5-year survival rate of children with ALL in most developed countries is 80–85% (4).
However, long-term antitumor treatment has a particularly large impact on children and there is still a high possibility of recurrence.
In total, 10–15% of pediatric patients will experience a relapse despite the successful induction of chemotherapy (5). Recurrence has been one of the key factors affecting the further enhancement of event-free survival rates and overall survival rates of children with ALL.
However, adoptive immunotherapy using genetic engineering to produce chimeric antigen receptor (CAR) T cells has shown unique advantages for the treatment of relapsed or refractory ALL (RR-ALL).
Treatment with CAR T-cells can alleviate refractory and secondary relapse of ALL to a certain extent, even in patients who fail to respond to bone marrow stem cell transplants (6,7).
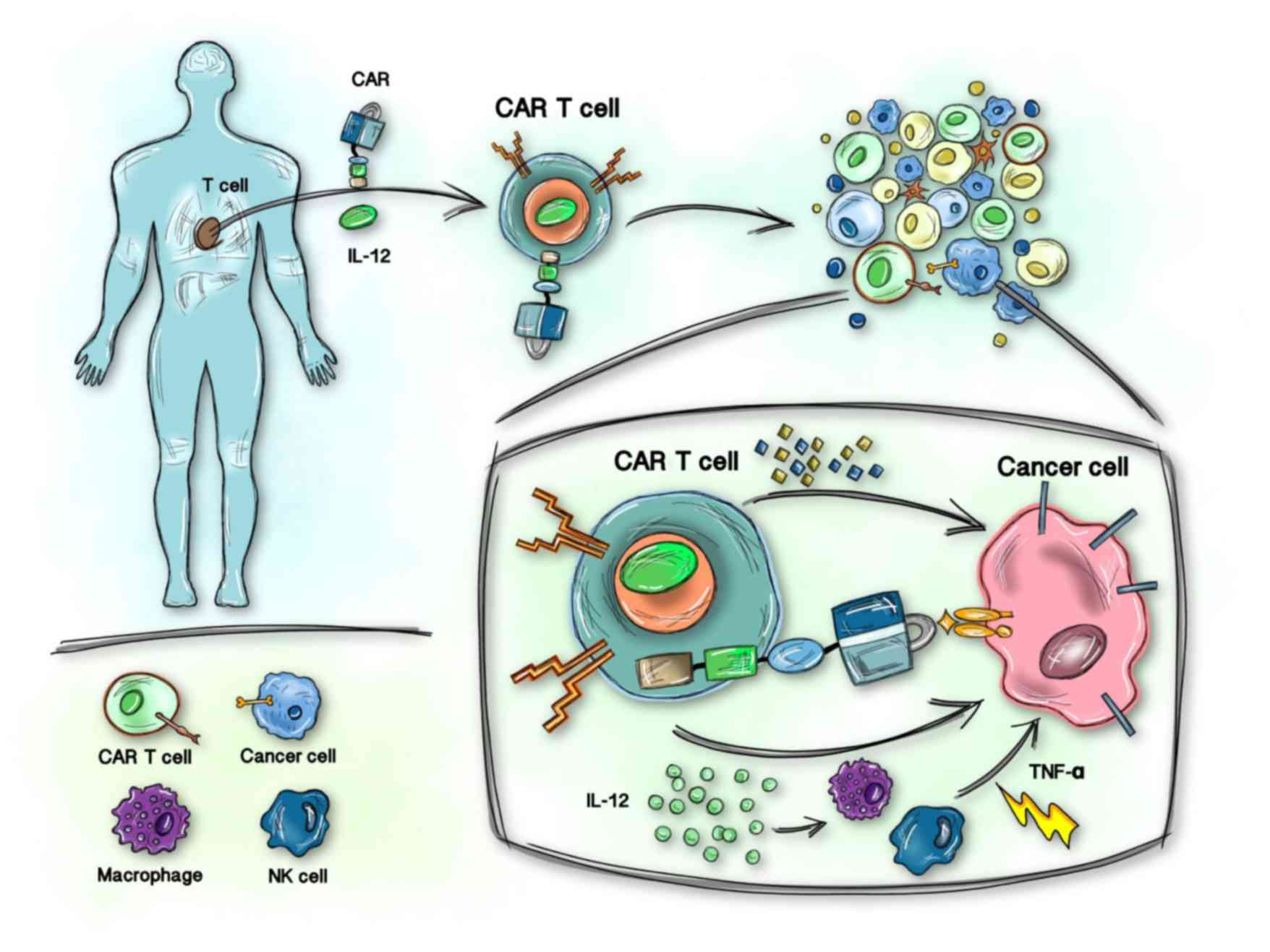
CAR T cells are modified normal T cells that are not bound by the major histocompatibility complex and have the ability to recognize tumor-associated antigens (TAAs) and kill tumor cells (8).
CAR T cells are superior to normal antibody-based drugs as they are comprised of autogenous or allogeneic living cells that can proliferate and develop in the body of a patient and remain stable for a considerable period of time. CAR T cells with cluster of differentiation (CD)19 (9,10), CD20 and CD22 (11,12) antigen-binding domains have been tested in humans; since CD19 is an essential biomarker of B cell lineage, CD19-CAR T cells are predominantly used to treat B-cell malignancies, including chronic lymphocytic leukemia, B-cell non-Hodgkin’s lymphoma and B-ALL (7,8).
In 2017, tisagenlecleucel, which is mainly used to treat patients <25 years of age with B-cell precursor ALL that is refractory or in secondary or later relapse stages, became the first CAR T-cell therapy approved by the US Food and Drug Administration (FDA) (13). In China, certain clinical trials being performed for RR-ALL also include this cellular immunotherapy (14–19).
Although CAR T-cell therapy has been recognized by major cancer medical centers, it still has serious side effects and toxicity, which can rapidly lead to fatal cytokine-release syndrome (CRS) (20,21) and neurotoxicity CAR T cell-related encephalopathy syndrome (CRES). Long-term side effects include a risk of chronic B-cell deficiency (14).
Hence, in the present review, the research progress and improvements in CAR T-cell treatment for ALL in children are assessed, and the relapse-related experiences and future prospects are analyzed.
CAR T cells
The main structure of the CAR protein comprises an intracellular signaling domain, a transmembrane domain, and an extracellular antigen-binding domain, usually derived from the single-chain variable fragment (scFv) targeted to human TAAs such as CD19 (23,24).
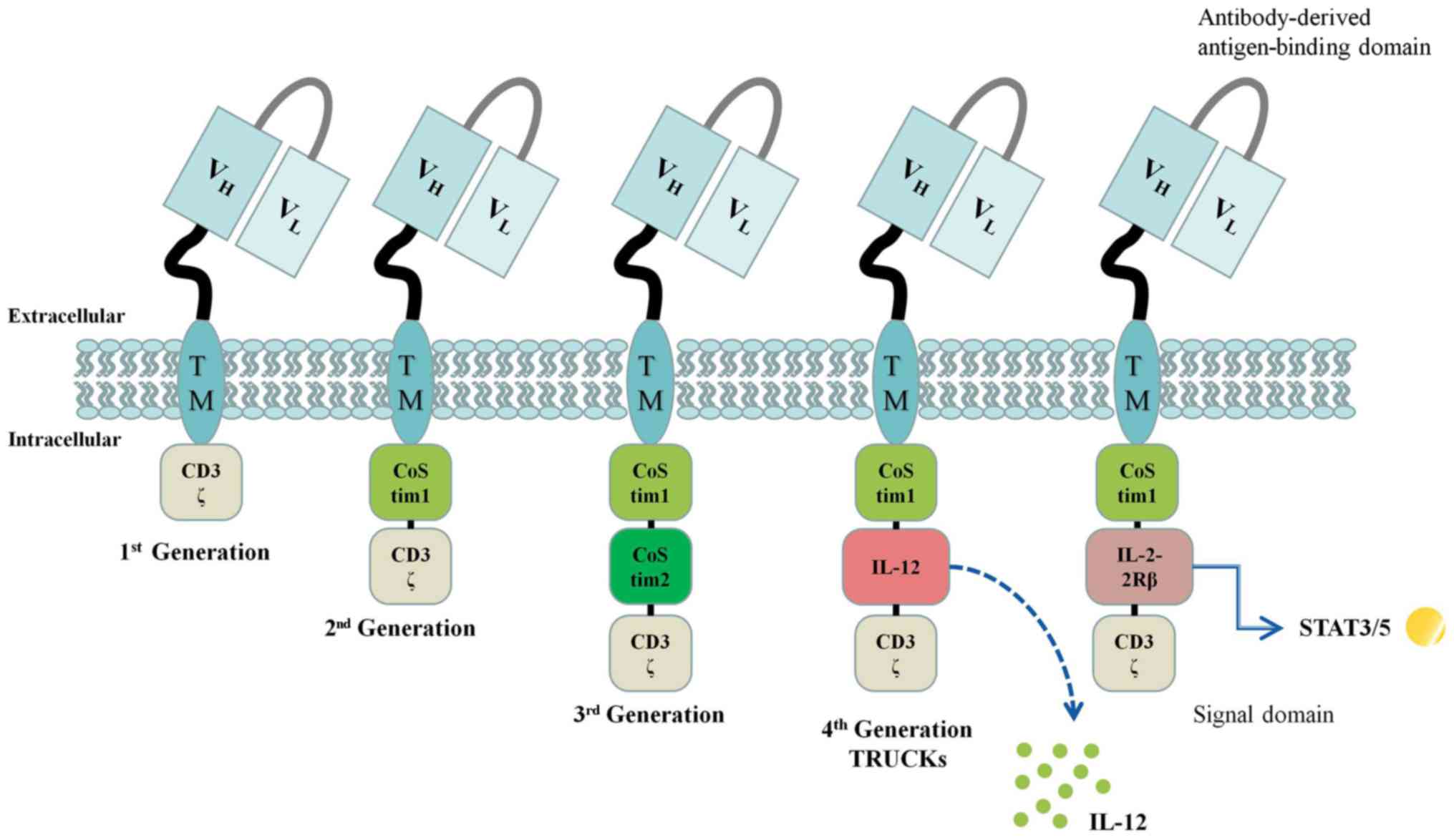
The intracellular signal domain is dominated by the T cell receptor CD3-ζ, as well as one (second-generation CAR) or two (third-generation CAR) connected co-stimulatory molecules, or alternatively, no co-stimulatory molecules (first-generation CAR) (Fig. 1).
Presently, second-generation CAR T cells are widely used in clinical practice, and the stability and killing effect of these cells are much greater than those of first-generation agents (25). However, current clinical results show that the third-generation agents possess no benefits above second-generation drugs (3,26).
Some scholars have developed the concept of T cells redirected for universal cytokine-mediated killing (TRUCKs), fourth-generation CARs (27). TRUCKs mediate CAR T-cell delivery of transgenic products [such as interleukin (IL)-12] to targeted tumor tissue to modulate the T cell responses (Fig. 2).
Fifth-generation CAR T cell products incorporate a novel co-stimulatory domain to activate other signaling pathways, such as the IL-2-2Rβ intracellular binding domain of signal transducer and activator of transcription 3/5 (25).
The selection of the target antigen is a key determinant of the specificity and effectiveness of the CAR and the safety of the genetically modified T cells. CD19 is specifically expressed by B-lymphocyte lineage cells at different stages of differentiation, and >95% of B-lymphoblastic leukemia cases express the CD19 antigen (28). To date, the CD19 antigen is the most widely used CD for CAR T-cell production.
CAR T cells designed using genetic engineering have been amplified in vitro and then transferred back into the patient without graft-versus-host disease. The CAR protein gives T cells the ability to recognize tumor antigens in a human leukocyte antigen-independent manner (29). Therefore, cytotoxic T cells can be activated in a short time and cytokines can be released to kill malignant cells.
Therapeutic effect of CD19-CAR T cells
Recently, CAR T cells that recognize and eliminate specific cancer cells have increased the recognition of their therapeutic usefulness, especially for hematological tumors. Table I summarizes some clinical trials (14,30–33) in which the rate of complete remission was unexpected positive for patients with ALL or RR-ALL.
CAR T-cell therapy is a good strategy to completely alleviate ALL and may be a novel strategy for RR-ALL. Previously, the available options were to increase the chemotherapy dose or change to different chemotherapy agents and regimens, which could put patients into remission; however, this was associated with a high recurrence rate (34,35), even with allogeneic hematopoietic stem cell transplantation (alloHSCT), which is also limited by the availability of suitable matched donors and potential immunologic complications (36).
Therefore, CAR T-cell therapy appears to be an extremely effective adoptive therapy that serves as a feasible, safe and efficacious approach to treat ALL, and particularly RR-ALL.
Table I. – Summary of reported therapy in trials of CAR T cells for children with ALL.
| First author, year | Patients | CAR T cells/kg | Therapeutic effect | CRS and CRES | Reported relapse | (Refs.) |
|---|---|---|---|---|---|---|
| Lee et al, 2015 | 21 children and young adults (aged 1–30 years), CD19+B-ALL (n=17) or NHL (n=4) | 0.028–3×106 cells | CR: 71% (12/17) | CRS (16/21; 76%), neurotoxicity (6/21; 29%) | Unknown | (30) |
| Maude et al, 2018 | 75 children and young adults (aged 3–23 years) CD19+ALL | 0.2–5.4×106 cells | CR: 81% (61/75) | CRS (58/75; 77%), neurological event (30/75; 40%) | 36% (22/61) relapsed (15 CD19- recurrence) | (31) |
| Gardner et al, 2017 | 45 children and young adults (aged 1.3–25.4 years), RR-CD19+ALL | 0.5/1/5/10×106 cells | CR: 93% (40/43) | 45% (18/40) relapse CRS (40/43; 93%), CRES (21/43; 49%) | (30) (7 CD19- recurrence) | |
| Zuo et al, 2019 | 48 children (aged 3–17 years), RR-CD19+ALL | (0.61–10.94)×106 cells | CR: 77% (37/48) | CRS (34/48; 71%), CRES (10/48; 21%) | 68% (25/37) relapse | (33) |
| Ma et al, 2019 | 10 pediatric patients with CD19 + RR B-ALL | (0.3–1.58)×106 cells | CR: 80% (8/10) | CRS (4/10; 40%), CRES (3/10; 30%) | 50% (4/8) relapse (3 CD19- recurrence) | (14) |
[i] CAR T, chimeric antigen receptor T; CRS, cytokine-release syndrome; CRES, CAR T cell-related encephalopathy syndrome; NHL, non-Hodgkin lymphoma; CR, complete remission; RR, relapsed or refractory; CD, cluster of differentiation; ALL, acute lymphoblastic leukemia.Table I. – Summary of reported therapy in trials of CAR T cells for children with ALL.
Furthermore, cytokines mediated by CAR T cells also play a prominent role in activating their antitumor effects; however, excessive cytokine levels can be life threatening (37). Patients receiving treatment should undergo continuous vital sign monitoring, as well as assessment of cytokine, tumor necrosis factor (TNF)-α, IL-6 and C-reactive protein (CRP) levels (6).
However, TNF-α and IL-6 are not typically monitored in the normal clinical setting. CRS should be highly suspected when the following signs occur (38), especially in the CRS-risk period after CAR T-cell infusion:
i) Fever ≥38°C;
ii) hypotension ≤90 mmHg for patients >10 years old;
iii) hypoxia with an arterial oxygen saturation of <90% when breathing room air; and iv) evidence of organ toxicity by the most recent Common Terminology Criteria for Adverse Events version 5.0 (CTCAE v5.0) grading system (38).
A clearer understanding of CRS has led to the publication of more detailed and standardized classifications and treatment management systems (21,38).
Although these experimental data are encouraging, there are obviously significant differences in pretreatment schemes, cellular injection doses (using various products) and even receptor composition among various medical centers.
It is thus important to reasonably summarize multi-center clinical trials and evaluate the most comprehensive treatment scheme statistically. The latest management guidelines of 2018, established by the MD Anderson Cancer Center and the HSCT Subgroup of the Pediatric Acute Lung Injury and Sepsis Investigators Network, comprehensively recommend that pediatric patients with B-cell precursor leukemias that are treatment refractory or in secondary or later relapse stages should receive CAR T-cell treatment, including nursing management, patient evaluation, examination before cell collection, attention to critical procedures when injecting cells and rescue measures, CRS monitoring and monitoring of CRES (38).
The guidelines also explicitly state that CAR T-cell therapy is a bridge to HCST as an interim therapy rather than a cure for ALL; therefore, the expectations might still be limited. Improvement in novel CAR T-cell therapy with enhanced efficacy and safety can potentially make it a mainstream cancer therapeutic.
Treatment-related complications
CAR T-cell therapy is associated with a series of side effects. Complications that require special attention in clinical practice include CRS, CRES, B-cell aplasia and palindromia. These adverse effects are some of the obstacles restricting the clinical application of CAR T-cell therapy and affecting its curative effect (39).
For example, while we use glucocorticoids with inhibitory effect to inflammatory responses to reverse CRS symptoms rapidly, glucocorticoids that are lymphocytotoxic will dampen expansion of the CAR T-cells in vivo and will affect therapeutic outcomes (40).
CRS
CRS occurs when cytokines are suddenly released in large quantities, leading to systemic inflammatory responses, including a high fever, increased levels of acute-phase proteins, and visceral and vascular endothelial damage, and eventually death from respiratory distress and heart failure (40,41).
As shown in Table I, numerous young adult and pediatric patients develop CRS after treatment with CD19-CAR T cells. Maude et al (31) conducted a global study on a cohort of tisagenlecleucel-treated pediatric and young adult patients with CD19+ B-cell RR-ALL. It was found that 77% of the patients in >25 medical centers involved in the trial developed CRS after tisagenlecleucel infusion, and almost half of these cases were life threatening, requiring intensive care (grades 3–4 CRS) (38,42).
Glucocorticoids that affect the proliferation and function of CAR T cells or anti-IL-6 therapy (e.g., tocilizumab; brand name, ACTEMRA; Genentech Inc.; Roche Diagnostics) can relieve CRS symptoms (21). More than half of patients with severe or life-threatening CRS exhibit resolution within 2 weeks of one or two doses of tocilizumab.
However, it has been demonstrated that patients with severe CRS are prone to early recurrence owing to the application of glucocorticoids (40). Therefore, in such patients, premature interventions after CAR T cells’ therapy may reduce the endurance/efficacy of T cells and decrease its therapeutic potential. Ultimately, the administration of timely and effective treatments to patients with severe CRS should be based on the rational/objective assessment of their clinical symptoms (such as high fever), and the timely monitoring of their biochemical indicators (such as CRP) and cytokine responses.
CRES
The serious neurotoxic symptoms associated with CAR T-cell therapy, known as CRES, usually present as headaches, emesis, tremors, delirium and seizures or cerebral edema (21,43). CRES is often associated with CRS or occurs after the fever and other CRS symptoms subside (42).
After CRS improves, neurotoxic encephalopathy can also improve. Although there is no exact pathophysiological explanation, evidence shows that this phenomenon is related to increased cytokines in the cerebrospinal fluid (21,44). Hu et al (43) first reported the case of a patient with CRS and neurotoxic symptoms (CRES) who improved after the reduction of intracranial pressure and glucocorticoid treatment, suggesting that the CRS-induced release of cytokines with a resultant overload might be one of main causes of neurotoxicity.
Moreover, the use of anti-IL-6 therapy seems to be more effective for CRES that occurs concurrently with CRS (42). Notably, soon after CRES onset, adult patients often have diminished attention, stuttering or writing dysfunction (42).
These signs may help us identify CRES patients as early as possible; therefore, the CARTOX 10-point neurological assessment tool or the Immune Effector Cell-Associated Encephalopathy (ICE) scoring system should be used, to evaluate potential acute neurological deficits due to CAR-T cell therapy in these adult patients (42,45–47).
However, symptoms in pediatric patients are subtle and completely different from those in adults, and the symptoms of early CRES are difficult to detect and diagnose in a timely manner in this population. The Cornell Assessment of Pediatric Delirium (CAPD) is an indispensable screening tool for the recognition of early CRES among young children and juveniles (<21 years of age, especially for patients less than 12 years old, the sensitivity and specificity are relatively high), as a CAPD score >8 represents delirium.
Furthermore, CAPD can also be used as a marker of CRES severity. Grade 2 CNS toxicities, including somnolence, confusion, encephalopathy, dysphasia, seizure (brief generalized seizure), and/or tremor, neurological assessment scores of 3–6 and a CAPD score <9 is indicative of grade 2 CRES. Conversely, a CAPD score >9 indicates grade 3–4 CRES.
If there is a risk of neurological symptoms, patients should be closely monitored for signs and symptoms of cerebral edema, for example, by specialized screening for papilloedema (38,47–49).
B-cell aplasia
scFv-based CARs only attack target specific antigens (such as CD19), which can be expressed by both tumor and normal cells, but in different amounts. Thus, on-target/on-tumor activity leads to a therapeutic effect, whereas an on-target/off-tumor effect induces a toxic reaction (50).
During long-term follow-up of patients receiving CAR T-cell therapy, one common adverse reaction is that CAR T cells attack normal B cells leading to B-cell lysis with a reduced immune response (38,51). In a clinical trial (52) using CAR T-cell therapy to treat refractory relapsed chronic lymphocytic leukemia, it was reported that all patients achieving complete remission developed B-cell aplasia, which lasted for up to 4 years in some patients due to the persistence of CAR T cells in their bodies.
Maude et al (6) observed that of 25 children with relapsed ALL, each had B cell aplasia, but none had serious infections due to B-cell aplasia. It is worth noting that by monitoring patients for the development of B-cell aplasia, the function of CAR T-cell therapy can be measured.
Such patients are managed with intravenous immunoglobulin repletion, which is only a temporary alternative treatment, until the B cells are rejuvenated. Selecting appropriate tumor-specific antigens and gradually increasing the injection dose could reduce the risk of missing the target (6,53,54).
It has also been reported that the introduction of suicide genes (55) and multi-antigen chimeric receptors (56) is an effective strategy. The most well-known approach is the fusion of the pro-apoptotic protein caspase-9 with the iCasp9 domain. When dimers of small molecules such as AP 1903 are introduced, the FKBP12 domain of iCasp9 becomes dimerized, resulting in the rapid apoptotic death of T cells (57).
Other complications
Acute tumor lysis syndrome is caused by the massive degradation of tumor cells with a rapid release of intracellular substances (such as potassium ions, phosphorus and nucleic acids). When the level of these intracellular substances exceeds the threshold of liver and kidney metabolic capacity, abnormal metabolism and electrolyte disorders of the body occur, leading to life-threatening severe arrhythmia and renal failure (58).
Allopurinol has been the main treatment for tumor lysis syndrome, and rasburicase is administered to manage serum uric acid levels (55). Moreover, hemophagocytic lymphohistiocytosis (HLH) is a serious and potentially fatal complication caused by the excessive secretion of inflammatory factors through the non-malignant proliferation of lymphocytes and histiocytes (59).
Secondary HLH is often referred to as macrophage-activation syndrome (MAS) (60). Given the overlapping symptoms of the inflammatory state, the clinical diagnosis of CAR T cell-related HLH (or MAS) and CRS-CRES is a great challenge (21,42).
The recommended diagnostic criteria for this rare situation have, however, been provided in the management guidelines for pediatric patients receiving CAR T-cell therapy, as described by Mahadeo et al (38).
Recurrence after CAR T-cell therapy
Although the remission rate of ALL in children is increasing gradually due to improved treatment strategies, the recurrence rate is still extremely high. Factors such as the short maintenance time of CAR T cells, insufficient copy number and antigen escape contribute to the recurrence of ALL after CAR T-cell therapy (61–63).
The detection or absence of the B-cell marker CD19 can be used to categorize recurrence as CD19-positive and CD19-negative leukemia, with the latter being more challenging to address. Antigen deletion is a notable phenomenon wherein tumor cells mutate the antigen gene to inhibit its expression (64) or induce defects in transcription factors involved in its synthesis to avoid death.
Ruella et al (65) encountered a case in which CAR, originally modified T cells, were bound to leukemia cells and proliferated in the body, eventually causing a CD19-negative relapse. CAR genes reversely bind to leukemia cells covering CD19 epitopes when CAR T cells are produced, allowing leukemia cells to mask CD19 proteins and develop CAR T-cell resistance to evade cellular immunity (65).
There is also the perspective that patients with RR-ALL express CD19-positive and -negative malignant cells. When CD19-CAR T cells kill malignant cells with the CD19 antigen, a few previously existing CD19-negative cells begin to take advantage of the cloning (9,66). Furthermore, CD19-negative relapse is resistant to CD19-CAR T-cell therapy, which can only be discontinued or switched to other clinical protocols (67,68).
Moreover, some patients relapse without antigen deletion. Monitoring the quantity of CAR T cells in patients reveals no detectable CAR T cells in the peripheral blood before relapse, which may be related to the insufficient copy number of CAR T cells and their inability to persist in the body for a long time (66,69).
Thus, the attending physician tends to administer a second injection of CAR T cells or an increased dose of the cells to prevent recurrence. However, the actual effect of this is minimal and could even lead to a poor prognosis (70). Hamieh et al (71) found that the CD19 antigen is transferred to CAR T-cells from malignant cells by trogocytosis, which not only leads to escape of the CD19 antigen, but also causes CAR T cells to kill each other and accelerates the depletion of T cells.
To weaken mutual killing, CAR T cells gradually express and activate T cell-inhibiting molecules such as programmed cell death protein-1, lymphocyte activation gene-3, and T-cell immunoglobulin and mucin domain 3, leading to the immune escape of tumors (71–73).
Similar experiments performed on CD22-CAR T cells, co-cultured with various cell lines such as SUP-B15 and Raji, and primary tumor cells from patients, also showed similar results that is CAR-induced trogocytosis was observed (71,74,75). This indicates that trogocytosis leads to tumor antigen deletion and metastasis via a universal mechanism that causes tumor surface antigen density to decrease after CAR T-cell therapy.
Even considering the advances in the field, such as the use of intensive chemotherapy, new targeted drugs and allogeneic hematopoietic stem cell transplantation, the remission rate of R/R ALL is still not negligible. Although CAR T-cell treatments lead to relatively controllable adverse events in R/R ALL patients, these patients still have a high risk of relapse (6,7). In such patients, CAR T-cell therapy could be a bridge for HSCT, providing temporary molecular remission for transplantation and improving the prognosis (76).
Strategies to prevent recurrence after CAR T-cell therapy
Dual antigen-expression construct
To prevent CD19-negative relapse, designing and using CAR T cells with bispecific antigen targets is a good strategy. Zah et al (56) designed a CDl9/CD20 bispecific CAR capable of OR-gate signal processing, which triggers robust T-cell responses as long as the target cells express either CD19 or CD20.
The optimized tandem-CARs control CD19-negative mutants, improving the killing ability of B-lineage malignant cells and reducing the recurrence rate. An increased disease burden is more likely to cause severe CRS and treatment has to be terminated, leading to recurrence.
It has been reported that this tandem CAR produces fewer cytokines than CD20-CAR, but is similar to that with CD19-CARs (56). Schneider et al (77) opined that tandem CARs are equally effective in standard disease models against single antigen-specific CARs, and might be more effective and less toxic with a high disease burden.
This could be attributable to the optimized cell killing with more moderate cytokine production. Furthermore, a dual CAR-expressing construct that combined CD19- and CD123-mediated CAR T cells demonstrated superior activity against B-ALL compared to that with single-expressing CAR T cells in vivo (78).
Co-stimulatory molecules
An insufficient copy number of CD19-CAR T cells would greatly curb the duration of action and thereby affect the therapeutic effects. Modification of the co-stimulatory molecule in the CAR signal domain structure could promote the expansion of CAR T cells and prolong their duration in vivo (68). The second- and third-generation CAR T cells are designed to contain two different co-stimulatory domain receptors; one is the CAR that provides the T-cell activation signal and the other is the chimeric co-stimulatory receptor. CD28 or 4-1BB (CD137) domain usually provide a costimulatory signal (30,40).
Lymphodepletion chemotherapy
CRS is known to severely affect CAR T cells with respect to the elimination of malignant cells. Ruella et al (79) found that the kinase inhibitor ibrutinib reduces CD19-CAR T-mediated CRS by inhibiting the production of inflammatory cytokines, without impairing the proliferation of T cells. In addition, 5-aza-2′-deoxycytidine (80), fludarabine (81), cyclophosphamide (10) and other drugs are also believed to improve the expansion and persistence of adoptive CAR T cells to enhance their anticancer efficacy.
Various combinations of these drugs constitute the lymphodepletion regimen. Most pediatric patients who participated in the tisagenlecleucel trial were initiated on a lymphodepletion regimen prior to CAR T-cell injection (31).
Conclusions and perspectives
After several decades of development, CAR T-cell design has become more sophisticated and its clinical application has become more extensive, even for the treatment of solid tumors. Moreover, a breakthrough occurred when the FDA approved tisagenlecleucel for the treatment of B-ALL, which has been gradually recognized and recommended by the global medical community, and this avant-garde technology could become the mainstream treatment for patients with hematologic tumors in the near future.
Professional guidelines for the management of CAR T-cell therapy in pediatric patients have also been issued. The multi-disciplinary nature of the treatment team ensures a complete understanding of relevant issues, and timely response strategies are instituted in the event of corresponding adverse reactions.
Problems encountered in the clinic currently suggest that improving the efficacy and safety of CAR T cells is an important starting point for CAR design. Dual-targeted CAR potentially reduces the risk of off-target effects, co-stimulatory molecules can stimulate the proliferation of T cells in vivo and suicide gene intervention is a powerful strategy to control adverse reactions.
However, a number of issues remain unresolved. First, the present review did not specify the range of doses of CAR T cells to be administered, given that a core strategy to minimize adverse reactions is required to obtain the maximum value with the minimum individual dose. Second, in the search for specific antigens for individual patients, there are still unresolved issues associated with relapse after CD19-CAR T-cell treatment.
Furthermore, it is debatable whether bone marrow transplantation should be continued or if CAR T-cell therapy should be repeated after the first CAR T-cell therapy. Finally, the exorbitant cost of tisagenlecleucel (with a one-time infusion cost of US$475,000) is also an economic challenge that has to be addressed to make this clinical treatment popular.
reference link : https://www.spandidos-publications.com/10.3892/ol.2020.11897?text=fulltext
More information: A.K. Park el al., “Effective Combination Immunotherapy using Oncolytic Viruses to Deliver CAR Targets to Solid Tumors,” Science Translational Medicine (2020). stm.sciencemag.org/lookup/doi/ … scitranslmed.aaz1863



{kind=link}