










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Experiments led by researchers at the Department of Energy’s Oak Ridge National Laboratory have determined that several hepatitis C drugs can inhibit the SARS-CoV-2 main protease, a crucial protein enzyme that enables the novel coronavirus to reproduce.
Inhibiting, or blocking, this protease from functioning is vital to stopping the virus from spreading in patients with COVID-19. The study, published in the journal Structure, is part of efforts to quickly develop pharmaceutical treatments for COVID-19 by repurposing existing drugs known to effectively treat other viral diseases.
“Currently, there are no inhibitors approved by the Food and Drug Administration that target the SARS-CoV-2 main protease,” said ORNL lead author Daniel Kneller.
“What we found is that hepatitis C drugs bind to and inhibit the coronavirus protease. This is an important first step in determining whether these drugs should be considered as potential repurposing candidates to treat COVID-19.”
The SARS-CoV-2 coronavirus spreads by expressing long chains of polyproteins that must be cut by the main protease to become functional proteins, making the protease an important drug target for researchers and drug developers.
In the study, the team looked at several well-known drug molecules for potential repurposing efforts including leupeptin, a naturally occurring protease inhibitor, and three FDA-approved hepatitis C protease inhibitors: telaprevir, narlaprevir and boceprevir.
The team performed room temperature X-ray measurements to build a three-dimensional map that revealed how the atoms were arranged and where chemical bonds formed between the protease and the drug inhibitor molecules.
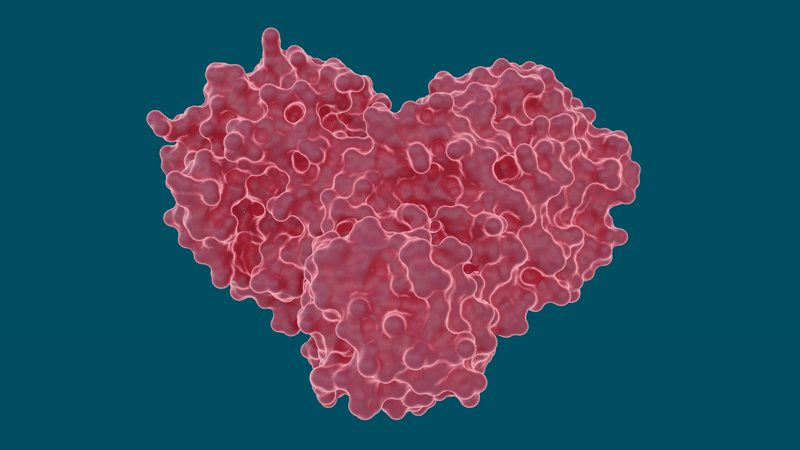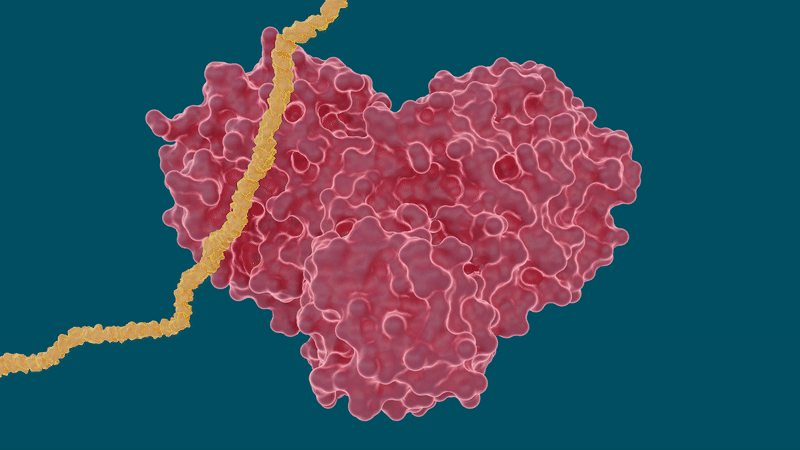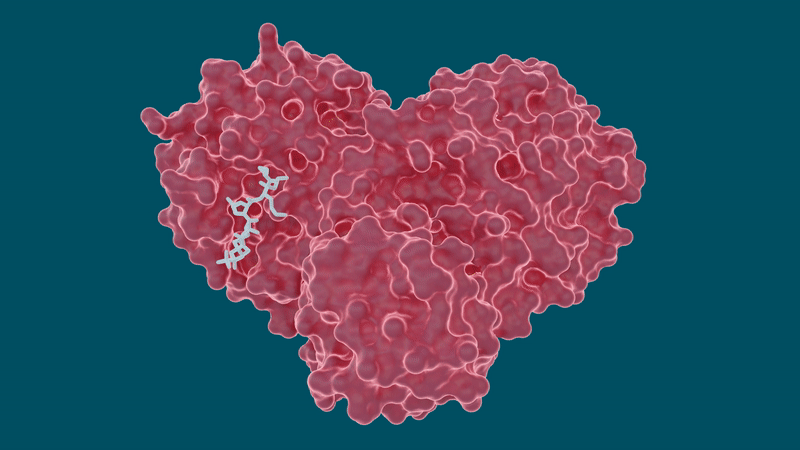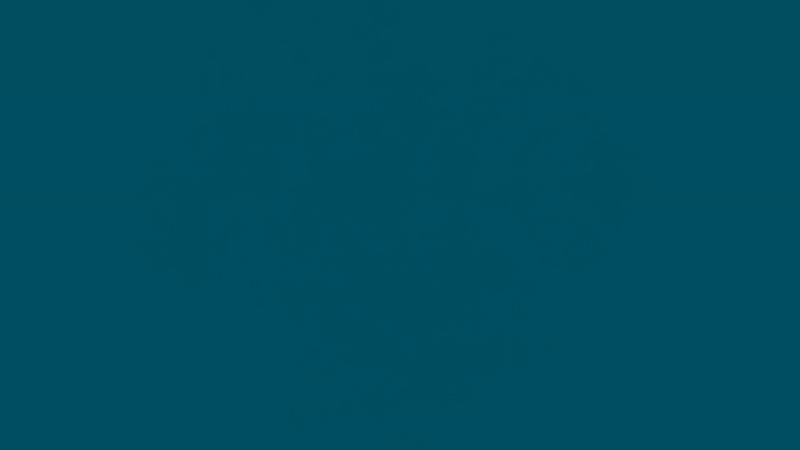
The experiments yielded promising results for certain hepatitis C drugs in their ability to bind and inhibit the SARS-CoV-2 main protease – particularly boceprevir and narlaprevir.
Leupeptin exhibited a low binding affinity and was ruled out as a viable candidate.
To better understand how well or how tightly the inhibitors bind to the protease, they used in vitro enzyme kinetics, a technique that enables researchers to study the protease and the inhibitor in a test tube to measure the inhibitor’s binding affinity, or compatibility, with the protease.
The higher the binding affinity, the more effective the inhibitor is at blocking the protease from functioning.
“What we’re doing is laying the molecular foundation for these potential drug repurposing inhibitors by revealing their mode of action,” said ORNL corresponding author Andrey Kovalevsky.
“We show on a molecular level how they bind, where they bind, and what they’re doing to the enzyme shape. And, with in vitro kinetics, we also know how well they bind. Each piece of information gets us one step closer to realizing how to stop the virus.”
The study also sheds light on a peculiar behavior of the protease’s ability to change or adapt its shape according to the size and structure of the inhibitor molecule it binds to. Pockets within the protease where a drug molecule would attach are highly malleable, or flexible, and can either open or close to an extent depending on the size of the drug molecules.
Before the paper was published, the researchers made their data publicly available to inform and assist the scientific and medical communities. More research, including clinical trials, is necessary to validate the drugs’ efficacy and safety as a COVID-19 treatment.
“The research suggests that hepatitis C inhibitors are worth thinking about as potential repurposing candidates.
Immediately releasing our data allows the scientific community to start looking at the interactions between these inhibitors and the protease,” said ORNL corresponding author Leighton Coates.
“You can’t design a drug without knowing how it works on a molecular level, and the data we’re providing is exactly what developers need to design stronger, more tightly binding drugs for more effective treatments.”
Orally available anti-coronavirus drugs would be highly desirable, as they can be given in an outpatient setting, i.e. without requiring hospitalization. As such they can be given upon diagnosis, which could prevent progression to more severe or protracted disease and thereby greatly reduce morbidity and mortality.
However, the only antiviral drug with proven efficacy against SARSCoV2, remdesivir, is an intravenous drug that can only be administered to hospitalized patients.
Even then it only reduces disease duration and risk of death by ∼30% each (3).
Dexamethasone has proven efficacy in improving survival in late-stage COVID-19 by suppressing the late hyperinflammatory responses that cause respiratory and organ failure rather than inhibiting viral replication (4). However, it is ineffective if given earlier (4), and even possibly harmful by inhibiting immune responses directed against the virus (5–7).
Thus, drugs that can effectively suppress coronavirus replication in an outpatient setting during the entire post-infection period do not exist, but are urgently needed.
Inhibitors of the coronavirus main protease (Mpro), a key protein in the coronavirus life cycle, could be especially effective as antiviral agents (8, 9).
Mpro (also called 3C-like protease due to its homology with enterovirus 3C protease) is a cysteine protease that cleaves the large nonstructural polyprotein of coronavirus into smaller pieces to assemble the RNA replicase (10).
Experimental inhibitors of coronavirus Mpro protect mice and cats from lethal coronavirus disease, validating Mpro as a medicinal target (11–13).
There is currently intense interest in discovering effective inhibitors for SARSCoV2 Mpro. As coronavirus Mpro species all share similar substrate specificity profiles (14), previously characterized inhibitors for other coronaviruses have, unsurprisingly, demonstrated inhibitory activity on SARSCoV2 Mpro as well. For example, the Michael acceptor compound N3, developed in 2005 as an inhibitor of SARSCoV1 Mpro (15), also inhibits the 96% identical SARSCoV2 Mpro in vitro (15, 16, 16).
A series of ketoamide-based inhibitors developed for SARSCoV1 Mpro were also effective against SARSCoV2 Mpro in vitro (17), and gave rise to the modified SARSCoV2 Mpro inhibitor 13b (18). Finally, GC376, a broad-spectrum aldehyde prodrug of various coronavirus Mpro species, and 11a, an aldehyde compound designed to inhibit SARSCoV1 Mpro were found to inhibit SARSCoV2 Mpro (19, 20).
However, it is not clear if known coronavirus Mpro inhibitors have the necessary attributes to be safe and effective oral therapeutics for human coronavirus diseases. In 11a and the active form of GC-376, an aldehyde reactive group (warhead) reacts with the active site cysteine, but aldehydes are generally considered non-ideal drug candidates due to toxicity caused by off-target reactions (21–24).
For example, the high reactivity of aldehyde warheads has been shown to drive non-specific inhibition of cysteine proteases (25). Protease inhibitors with ketoamide and Michael acceptor warheads can exhibit better specificity, stability, and oral bioavailability than those with aldehydes (26–29), and examples have progressed to late-stage clinical trials or regulatory approval (30, 31).
Compounds 13b and N3 utilize ketoamide and Michael acceptor warheads respectively, but they appear to have insufficient efficacy to be therapeutically useful.
While 13b demonstrates a 50% inhibitory concentration (IC50) on Mpro in vitro of 670 nM, the 50% effective concentration (EC50) in blocking SARSCoV2 replication in human cells is ∼4 μM (18).
This compares unfavorably to the achieved concentration in mouse lungs of <0.1 μM (18). For N3, while it demonstrates a low IC50 in vitro, its EC50 for SARSCoV2 replication is 17 μM (16), indicating poor cell permeability. This is not surprising given its large size (mw 681) and numerous polar groups. Thus, the development of new Mpro inhibitor designs with low toxicity and low EC50 in cells remains an urgent priority.
Multiple clinically approved protease inhibitors incorporate cyclic structures to improve affinity by pre-organizing the inhibitors into a conformation favorable for binding (32).
In energetic terms, this is considered paying the entropic penalty of conformational rigidification prior to binding (33, 34), and cyclization has been experimentally confirmed to increase protease inhibitor affinity (35).
In this study, we explore the ability of a cyclic moiety to improve the affinity of coronavirus Mpro inhibitors. We developed a novel inhibitor, ML1000, with molecular weight of 549 Da and IC50 value of 12 nM on SARSCoV2 Mpro.
ML1000 inhibits SARSCoV2 replication in human cells with an EC50 of 0.1 µM, and thus is the highest-potency non-aldehyde Mpro inhibitor reported so far. ML1000 represents a new structural class of potent ketoamide inhibitors of coronavirus Mpro, and may serve as a promising starting point for the development of drug candidates against human coronavirus diseases.
RESULTS
While visualizing the co-crystal structure of 13b and SARSCoV2 Mpro (18), we noticed that 13b adopts a pronounced kink in its main chain at the P2 residue, i.e. the 2nd residue N-terminal to the scissile bond facing the enzyme’s S2 pocket (Fig. 1A).
Due to our previous work with HCV protease and its small-molecule inhibitors (36), we recognized this kink to be similar to that created by proline analog rings in the clinically approved anti-HCV drugs boceprevir, narlaprevir, and telaprevir (Fig. 1B-D).
Like 13b, these drugs are ketoamide-based covalent inhibitors, serving as substrates for nucleophilic attack by the deprotonated active-site cysteine.

HCV protease inhibitors with a P2 proline analog can be docked into the SARSCoV2 Mpro active site. (A) Co-crystal structure of SARSCoV2 Mpro and inhibitor 13b (PDB 6Y2G). (B) Using Pymol, boceprevir was placed into the SARSCoV2 Mpro active site and unconstrained bonds were manually rotated for optimal complementary with the S1 and S2 pockets and hydrogen-bonding to the backbone carbonyl of Glu-166. (C) Telaprevir was similarly docked into the SARSCoV2 Mpro active site for optimal complementary with the S1, S2, and S4 pockets and hydrogen-bonding to the backbone carbonyl of Glu-166. (D) Alignment of the 13b-Mpro cocrystal with the manually docked boceprevir structure shows that the backbone of the P2-analogous segment of 13b is superimposable with the proline analog of boceprevir.
Manual rigid docking of boceprevir showed that it could fit into the active site of SARSCoV2 Mpro with good shape complementarity by its P1, P2, and P4 groups (Fig. 1B).
In addition, the urea group of boceprevir at the P3-P4 junction could engage in a bidentate hydrogen bond with the backbone carbonyl of Mpro Glu-166.
The boceprevir derivative narlaprevir also demonstrated complementary in its P1 and P2 groups, which are identical to boceprevir, but its P4 group was clearly too large for the S4 pocket. Telaprevir could also be manually docked with good complementarity of its P1 sidechain and of its P2 group, which is a different proline analog from that of boceprevir (Fig. 1C), whereas its P4 group appeared slightly too large for the S4 pocket.
The P3 group of coronavirus Mpro substrates face out into solution (37), as does the t-butyl group in the analogous position of boceprevir, telaprevir, and narlaprevir. Interestingly, the φ and Ψ angles of the proline ring in boceprevir precisely retraced the backbone atoms of 13b in the bound configuration (Fig. 1D).
We thus hypothesized that boceprevir and, to a lesser extent, narlaprevir and telaprevir, may be able to inhibit SARSCoV2 Mpro.
To address this hypothesis, we performed a preliminary rapid assessment using a live-cell assay of Mpro function. We co-expressed SARSCoV2 Mpro and a substrate protein comprising green fluorescent protein (GFP), a substrate site, and the membrane-targeting CAAX sequence (Fig. 2A).
Cleavage at the substrate sequence liberates GFP, allowing quantitation of activity by immunoblotting. We could then assess the ability of drugs to inhibit Mpro activity in cells. In HEK293A cells, we found boceprevir and telaprevir to inhibit SARSCoV2 Mpro at micromolar concentrations (Fig. 2B).
These results provided evidence of the ability of HCV inhibitors with a P2 proline group to inhibit SARSCoV2 Mpro.

Preliminary evidence of HCV protease inhibitor activity on SARSCoV2 Mpro. (A) Schematic of the protease reporter construct. An immature version of Mpro with reduced activity was used for this experiment to mimic multiple natural states of Mpro. During the experiment, Mpro is expected to self-activate by removing the terminal extensions. (B) Immunoblot of lysates from HEK293A cells transiently transfected with the reporter construct shown in (A) and grown in the absence or presence of different concentrations of telaprevir or boceprevir shows suppression of substrate cleavage at micromolar drug concentrations.
We next performed a pilot test of the ability of HCV inhibitors with a P2 proline group to inhibit SARSCoV2 Mpro in vitro (Fig. 3A).
As controls, we also tested GC-376 as a known broad-spectrum coronavirus Mpro inhibitor, ebselen and disulfiram as compounds recently reported to inhibit SARSCoV2 Mpro (16), and ritonavir as a protease inhibitor without any structural homology to coronavirus Mpro substrates. Indeed, we observed that boceprevir, narlaprevir, telaprevir, GC-376, disulfiram, and ebselen (at 150 μM concentration in air-equilibrated buffer) were able to inhibit SARSCoV2 Mpro, whereas ritonavir showed no inhibitory effect (Fig. 3A).
Incidentally, disulfiram and ebselen showed no inhibitory effect in the presence of DTT, whereas other compounds were unaffected by DTT. These results are consistent with proposals of disulfiram and ebselen forming a reducible bond with Mpro (38).
As the interior of the cell is reducing, reduction of ebselen adducts could contribute to its relatively high EC50 of 4.7 μM for blocking SARSCoV2 replication in cells (16).

We then performed full inhibition curves SARSCoV2 Mpro to obtain IC50 values for the above compounds. The initial pilot test above was performed with SARSCoV2 Mpro with a C-terminal His6-tag for rapid purification, but the presence of a C-terminal extension may have an inhibitory effect on protease activity (39, 40). We thus performed inhibition curves on both C-terminally extended and fully mature SARSCoV2 Mpro (Fig. 3B).
We tested drugs in the presence of DTT, except for ebselen and disulfiram, for which we omitted DTT. Our results showed that the proline-containing HCV protease inhibitors boceprevir, telaprevir, and narlaprevir all could inhibit activity to some extent in both C-terminally extended and fully mature forms of SARSCoV2 Mpro. Among these, boceprevir was the most potent, with an IC50 value of 4.1 μM on mature SARSCoV2 Mpro (Fig. 3B, Table 1).

Having established that the P2 proline analogues in boceprevir, telaprevir, and narlaprevir were compatible with binding to the SARSCoV2 Mpro active site, we next sought to design next-generation coronavirus inhibitors that could combine the entropic stabilization conferred by these P2 rings with side groups optimized for coronavirus Mpro binding. A P1 Gln residue is strongly preferred by all coronavirus Mpro species (14).
In fact, this preference is conserved with the related enterovirus 3C proteases such as human rhinovirus (HRV) protease and even with the more distantly related potyvirus proteases such as tobacco etch virus (TEV) protease (41, 42). Beginning with the HRV protease inhibitors AG7088 (rupintrivir) and its derivative AG7404 (43, 44), enterovirus and coronavirus protease inhibitors have incorporated a γ-lactam group to mimic the hydrogen bond acceptor function of Gln in the P1 position.
Enzyme-inhibitor cocrystal structures have confirmed that the γ-lactam amide group is positioned similarly to the Gln amide group in natural structures (37, 45). We thus designed a novel inhibitor, ML1000, with the ketoamide warhead of boceprevir, the γ-lactamyl P1 group of rupintrivir, and the P2 through P4 moieties (including backbone and side chains) of boceprevir (Fig. 4A).
We also designed a second inhibitor, ML1100, composed of the ketoamide warhead of boceprevir, the γ-lactamyl P1 group of rupintrivir, the P2 cyclic structure of telaprevir, and the P3 and P4 moieties of boceprevir (Fig. 4A).
We retained the P3 and P4 structure of boceprevir as these groups did not demonstrate any clashes with the enzyme pocket in the manually docked boceprevir model, and as their hydrophobicity likely contributes to membrane permeability.

Design and in vitro potency of a novel coronavirus Mpro inhibitors. (A) Structures of boceprevir, telaprevir, ML1000, and ML1100. ML1000 is essentially boceprevir with a γ-lactamyl group in place of the P1 cyclobutanyl group. ML1100 replaces the bicyclic P2 proline analog of boceprevir with that of telaprevir. (B) IC50 measurements of ML1000 and ML1100 with 100 nM mature SARSCoV2 Mpro. With enzyme concentration at 100 nM, the lowest possible IC50 that can be detected in theory is 50 nM. Thus, IC50 values were also measured for ML1000 and GC-376 with 20 nM enzyme. (C) IC50 values of ML1000 and GC-376 measured with 20 nM mature SARSCoV2 Mpro were 12 and 14 nM, respectively. Thus, ML1000 and GC-376 shows tight binding, even at 20 nM of Mpro, and the IC50 values may still be limited by the enzyme concentration. Mean values of 3 independent experiments are shown. Error bars represent standard deviation.
ML1000 and ML1100 proved to be highly potent inhibitors of SARSCoV2 Mpro. In the presence of 100 nM of enzyme, ML1000 and ML1100 produced IC50 values of 34 nM and 147 nM (Fig. 4B, Table 1), compared to 54 nM for GC-376 (Fig. 3B, Table 1).
Against the Mpro enzyme from the distantly related coronavirus mouse hepatitis virus (MHV), ML1000 and ML1100 exhibited IC50 values of 130 and 301 nM, respectively, compared to 67 nM for GC-376 (Table 1, Supporting Fig. 1).
These results demonstrate that ketoamide inhibitors with proline ring analogs at the P2 position can function as broad-spectrum coronavirus Mpro inhibitors with potency approaching that of aldehyde inhibitors.

We noted that the measured IC50 values of ML1000 and GC-376 for SARSCoV2 Mpro were within experimental error of the theoretical limit of half of the enzyme concentration in the assay (46), hindering the ability to discern differences in potency between them. We thus also measured IC50 values at a lower SARSCoV2 Mpro concentration of 20 nM (Fig. 4C).
In these conditions, ML1000 and GC-376 still demonstrated comparable IC50 values of 12 and 14 nM, respectively, below the concentration of Mpro (i.e. the tight binding limit).
Physiological temperature, compared to the 30 °C at which these assays are usually performed, could potentially increase structural flexibility and alter drug affinity of Mpro. However, we found no significant difference in potency between 37 °C and 30 °C for each inhibitor (Table 1, Supporting Fig. 2).
These results indicate that ML1000 has high potency in vitro and represents the tightest-binding non-aldehyde SARSCoV2 Mpro inhibitor discovered so far.

Next, we tested the efficacy of the new inhibitors against SARSCoV2 Mpro in human Huh7 cells by measuring the extent of cleavage of coexpressed substrate (Fig. 5). Here, we observed the rank order of effectivenss to be GC-376 > ML1000 > ML1100 > boceprevir (Fig. 5).
It should be emphasized that this assay does not directly measure the fraction of active Mpro. If a small fraction of active Mpro is sufficient to result in substrate cleavage, this would effectively increase the measured concentration needed to suppress protease activity.
However, the assay does provide an assessment of the relative ability of a set of drugs to cross the cellular membrane and inhibit Mpro within living cells.

Finally, we tested the ability of ML1000 and ML1100 to inhibit SARSCoV2 virus replication in Caco-2 human intestinal cells. For comparison, we tested boceprevir and GC-376. Boceprevir inhibited viral replication with an EC50 of 0.2 µM, while GC-376 was even more potent, with a remarkably low EC50 of 0.1 nM (Table 2, Fig. 6). ML1000 and ML1100 inhibited SARSCoV2 replication with EC50 values of 0.1 and 0.2 µM, respectively (Table 2, Fig. 6).
While the enhancement in anti-viral efficacy of ML1000 relative to boceprevir is not as large as the enhancement in enzyme inhibition observed in vitro (Table 1), the EC50 value of ML1000 is nevertheless the lowest reported to date for any non-aldehyde inhibitors of SARSCoV2 Mpro.
In addition, ML1000 and ML1100 exhibited no cytotoxicity at the highest dose tested of 100 μM in Caco-2 cells and Huh-7 cells (Table 2, Supporting Fig. 3), resulting in selectivity indices of >1000 and >500 respectively. These results confirm that ML1000 is a potent and selective ketoamide inhibitor of SARSCoV2 replication.



REFERENCES
- 1. M. Letko, S. N. Seifert, K. J. Olival, R. K. Plowright, V. J. Munster, Batborne virus diversity, spillover and emergence. Nat Rev Microbiol 18, 461–471 (2020). Google Scholar
- 2. W. H. Organization, Weekly Operational Update on COVID19, 28 August 2020. Google Scholar
- 3. J. H. Beigel et al., Remdesivir for the Treatment of Covid19 Preliminary Report. N Engl J Med (2020). Google Scholar
- 4. C. G. Recovery et al., Dexamethasone in Hospitalized Patients with Covid19 Preliminary Report. N Engl J Med (2020). Google Scholar
- 5. D. W. Cain, J. A. Cidlowski, After 62 years of regulating immunity, dexamethasone meets COVID19. Nat Rev Immunol (2020). Google Scholar
- 6.M. A. Lim, R. Pranata, Worrying situation regarding the use of dexamethasone for COVID19. Ther Adv Respir Dis 14, 1753466620942131 (2020). Google Scholar
- 7. T. C. Theoharides, P. Conti, Dexamethasone for COVID19? Not so fast. J Biol Regul Homeost Agents 34, (2020). Google Scholar
- 8. X. Deng et al., Coronaviruses resistant to a 3Clike protease inhibitor are attenuated for replication and pathogenesis, revealing a low genetic barrier but high fitness cost of resistance. J. Virol. 88, 11886–11898 (2014). Abstract/FREE Full TextGoogle Scholar
- 9. S. Ullrich, C. Nitsche, The SARSCoV2 main protease as drug target. Bioorg. Med. Chem. Lett. 30, 127377 (2020). Google Scholar
- 10. Y. Kim et al., Broadspectrum antivirals against 3C or 3Clike proteases of picornaviruses, noroviruses, and coronaviruses. J. Virol. 86, 11754–11762 (2012). Abstract/FREE Full TextGoogle Scholar
- 11. Y. Kim et al., Broadspectrum inhibitors against 3Clike proteases of feline coronaviruses and feline caliciviruses. J. Virol. 89, 4942–4950 (2015). Abstract/FREE Full TextGoogle Scholar
- 12. Y. Kim et al., Reversal of the Progression of Fatal Coronavirus Infection in Cats by a BroadSpectrum Coronavirus Protease Inhibitor. PLoS Pathog 12, e1005531 (2016). CrossRefPubMedGoogle Scholar
- 13. N. C. Pedersen et al., Efficacy of a 3Clike protease inhibitor in treating various forms of acquired feline infectious peritonitis. J Feline Med Surg 20, 378–392 (2018). CrossRefPubMedGoogle Scholar
- 14. C. P. Chuck, H. F. Chow, D. C. Wan, K. B. Wong, Profiling of substrate specificities of 3Clike proteases from group 1, 2a, 2b, and 3 coronaviruses. PLoS One 6, e27228 (2011). CrossRefPubMedGoogle Scholar
- 15. H. Yang et al., Design of widespectrum inhibitors targeting coronavirus main proteases. PLoS Biol 3, e324 (2005). CrossRefPubMedGoogle Scholar
- 16. Z. Jin et al., Structure of Mpro from COVID19 virus and discovery of its inhibitors. Nature 582, 289–293 (2020). Google Scholar
- 17. L. Zhang et al., αKetoamides as BroadSpectrum Inhibitors of Coronavirus and Enterovirus Replication: StructureBased Design, Synthesis, and Activity Assessment. J. Med. Chem. 63, 4562–4578 (2020). Google Scholar
- 18. L. Zhang et al., Crystal structure of SARSCoV2 main protease provides a basis for design of improved αketoamide inhibitors. Science 368, 409–412 (2020). Abstract/FREE Full TextGoogle Scholar
- 19. W. Dai et al., Structurebased design of antiviral drug candidates targeting the SARSCoV2 main protease. Science 368, 1331–1335 (2020). Abstract/FREE Full TextGoogle Scholar
- 20. W. Vuong et al., Feline coronavirus drug inhibits the main protease of SARSCoV2 and blocks virus replication. Nat Commun 11, 4282 (2020). Google Scholar
- 21. K. Gluza, P. Kafarski, Transition state analogues of enzymatic reaction as potential drugs. Drug Discovery (2013). Google Scholar
- 22. S. L. Harer, M. S. Bhatia, N. M. Bhatia, Proteasome inhibitors mechanism; source for design of newer therapeutic agents. J Antibiot (Tokyo) 65, 279–288 (2012). PubMedGoogle Scholar
- 23.M. Siklos, M. BenAissa, G. R. J. Thatcher, Cysteine proteases as therapeutic targets: does selectivity matter? A systematic review of calpain and cathepsin inhibitors. Acta Pharmaceutica Sinica B (2015). Google Scholar
- 24. Vasudevan et al., Covalent binders in drug discovery. Prog. Med. Chem. 58, 1–62 (2019). Google Scholar
- 25. L. Zhu et al., Peptide aldehyde inhibitors challenge the substrate specificity of the SARScoronavirus main protease. Antiviral Res 92, 204–212 (2011). CrossRefPubMedWeb of ScienceGoogle Scholar
- 26. D. G. Barrett et al., Orally bioavailable small molecule ketoamidebased inhibitors of cathepsin K. Bioorg. Med. Chem. Lett. 14, 2543–2546 (2004). PubMedGoogle Scholar
- 27. K. O. Chang, Y. Kim, W. C. Groutas, D. Hua, S. Lj, Broadspectrum antivirals against 3c or 3clike proteases of picornaviruslike supercluster: picornaviruses, caliciviruses and coronaviruses. US Patent 9,474,759 (2016). Google Scholar
- 28. Y. Kim et al., Potent inhibition of enterovirus D68 and human rhinoviruses by dipeptidyl aldehydes and αketoamides. Antiviral Res 125, 84–91 (2016). Google Scholar
- 29. Y. Shirasaki et al., Exploration of orally available calpain inhibitors 2: peptidyl hemiacetal derivatives. J. Med. Chem. 49, 3926–3932 (2006). PubMedGoogle Scholar
- 30. S. Amslinger, The tunable functionality of α, βunsaturated carbonyl compounds enables their differential application in biological systems. ChemMedChem (2010). Google Scholar
- 31. F. Sutanto, M. Konstantinidou, A. Dömling, Covalent inhibitors: a rational approach to drug discovery. RSC Medicinal Chemistry (2020). Google Scholar
- 32. D. I. Soumana et al., Structural and Thermodynamic Effects of Macrocyclization in HCV NS3/4A Inhibitor MK5172. ACS Chem Biol 11, 900–909 (2016). CrossRefGoogle Scholar
- 33. Z. Fang, Y. Song, P. Zhan, Q. Zhang …, Conformational restriction: an effective tactic in’followon’based drug discovery. Future Medicinal Chemistry (2014). Google Scholar
- 34. P. Y. Lam, P. K. Jadhav, C. J. Eyermann, C. N. Hodge …, Rational design of potent, bioavailable, nonpeptide cyclic ureas as HIV protease inhibitors. … (1994). Google Scholar
- 35. K. Ghosh, G. E. Schiltz, L. N. Rusere …, Design and synthesis of potent macrocyclic HIV1 protease inhibitors involving P1–P2 ligands. Organic … (2014). Google Scholar
- 36. C. L. Jacobs, R. K. Badiee, M. Z. Lin, StaPLs: versatile genetically encoded modules for engineering druginducible proteins. Nat Methods 15, 523–526 (2018). Google Scholar
- 37. M. F. Hsu et al., Mechanism of the maturation process of SARSCoV 3CL protease. J. Biol. Chem. 280, 31257–31266 (2005). Abstract/FREE Full TextGoogle Scholar
- 38. H. Sies, M. J. Parnham, Potential therapeutic use of ebselen for COVID19 and other respiratory viral infections. Free Radic Biol Med 156, 107–112 (2020). Google Scholar
- 39. T. Muramatsu et al., Autoprocessing mechanism of severe acute respiratory syndrome coronavirus 3Clike protease (SARSCoV 3CLpro) from its polyproteins. FEBS J 280, 2002–2013 (2013). CrossRefPubMedGoogle Scholar
- 40. X. Xue et al., Production of authentic SARSCoV M(pro) with enhanced activity: application as a novel tagcleavage endopeptidase for protein overproduction. J. Mol. Biol. 366, 965–975 (2007). CrossRefPubMedGoogle Scholar
- 41. M. J. Adams, J. F. Antoniw, F. Beaudoin, Overview and analysis of the polyprotein cleavage sites in the family Potyviridae. Mol Plant Pathol 6, 471–487 (2005). CrossRefPubMedWeb of ScienceGoogle Scholar
- 42. J. Tan et al., 3C protease of enterovirus 68: structurebased design of Michael acceptor inhibitors and their broadspectrum antiviral effects against picornaviruses. J. Virol. 87, 4339–4351 (2013). Abstract/FREE Full TextGoogle Scholar
- 43. D. A. Matthews et al., Structureassisted design of mechanismbased irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc. Natl. Acad. Sci. U. S. A. 96, 11000–11007 (1999). Abstract/FREE Full TextGoogle Scholar
- 44. K. Patick et al., In vitro antiviral activity and singledose pharmacokinetics in humans of a novel, orally bioavailable inhibitor of human rhinovirus 3C protease. Antimicrob. Agents Chemother. 49, 2267–2275 (2005). Abstract/FREE Full TextGoogle Scholar
- 45. S. Yang et al., Synthesis, crystal structure, structureactivity relationships, and antiviral activity of a potent SARS coronavirus 3CL protease inhibitor. J. Med. Chem. 49, 4971–4980 (2006). CrossRefPubMedGoogle Scholar
- 46. R. Z. Cer, U. Mudunuri, R. Stephens, F. J. Lebeda, IC50toKi: a webbased tool for converting IC50 to Ki values for inhibitors of enzyme activity and ligand binding. Nucleic Acids Res. 37, W441–5 (2009). CrossRefPubMedGoogle Scholar
- 47. B. R. Bacon et al., Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med 364, 1207–1217 (2011). CrossRefPubMedWeb of ScienceGoogle Scholar
- 48. L. Fu et al., Both Boceprevir and GC376 efficaciously inhibit SARSCoV2 by targeting its main protease. Nat Commun 11, 4417 (2020). Google Scholar
- 49. C. Ma et al., Boceprevir, GC376, and calpain inhibitors II, XII inhibit SARSCoV2 viral replication by targeting the viral main protease. Cell Res 30, 678–692 (2020). Google Scholar
- 50. B. J. Anson et al., Broadspectrum inhibition of coronavirus main and papainlike proteases by HCV drugs. researchsquare.com (2020). Google Scholar
- 51. X. Zuo et al., Expression and purification of SARS coronavirus proteins using SUMOfusions. Protein Expr Purif 42, 100–110 (2005). CrossRefPubMedGoogle Scholar
More information: Daniel W. Kneller et al, Malleability of the SARS-CoV-2 3CL Mpro Active-Site Cavity Facilitates Binding of Clinical Antivirals, Structure (2020). DOI: 10.1016/j.str.2020.10.007



{kind=link}