










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Il dogma centrale della biologia molecolare afferma che il DNA viene trascritto in mRNA, che viene successivamente tradotto in proteina [8]. Il flusso di informazioni genetiche nel tempo e nello spazio è orchestrato da complessi meccanismi di regolazione. La terapia genica rappresenta l’introduzione di materiale genetico nelle cellule e nei tessuti biologici di un individuo.
Tecniche come l’inserimento, l’alterazione o la rimozione di geni sono impiegate per correggere i geni difettosi responsabili dello sviluppo della malattia, che poi cura una malattia o migliora lo stato clinico di un paziente [9].
Diversi vettori sono stati utilizzati per la terapia genica e sono generalmente classificati come vettori non virali e virali. I vettori non virali possiedono diversi vantaggi rispetto ai vettori virali, tra cui una bassa immunogenicità dell’ospite e un potenziale di scale-up [10].
Tuttavia, il successo della terapia genica non virale è stato molto limitato in passato, principalmente a causa delle barriere esistenti per la consegna del DNA plasmidico (pDNA), ad esempio, la necessità di attraversare la membrana nucleare prima della traduzione, la presenza di geni di resistenza agli antibiotici nel pDNA e, soprattutto, la difficoltà nel controllare e regolare l’espressione a lungo termine.
La mancanza di controllo dell’espressione a lungo termine del pDNA rappresenta un enorme svantaggio in termini di durata del trattamento e possibili effetti collaterali, che è in contrasto con i farmaci convenzionali, dove il trattamento può essere interrotto istantaneamente. Questi svantaggi del pDNA possono essere eventualmente superati utilizzando mRNA [11].
L’mRNA trasporta le informazioni genetiche dal DNA nel nucleo al citosol, dove viene utilizzato dai ribosomi come modello per la sintesi proteica. A differenza del pDNA, l’mRNA è efficace nelle cellule mitotiche e non mitotiche perché l’mRNA esercita la sua funzione nel citoplasma, quindi la sua funzione non dipende dalla divisione cellulare attiva.
Inoltre, a differenza del pDNA o dei vettori virali, l’mRNA non contiene geni estranei aggiuntivi, il che rende l’mRNA un vettore più sicuro. La sfida dell’espressione a lungo termine rappresentata dal pDNA può essere superata anche utilizzando mRNA, poiché l’mRNA media un’espressione rapida e transitoria della proteina codificata e la durata della produzione è ben definita (di solito pochi giorni o settimane, a seconda sulla specifica piattaforma mRNA).
Ciò rende l’espressione dell’mRNA più facile da controllare rispetto all’espressione genica da pDNA e vettori virali [11]. Inoltre, la produzione di mRNA è priva di cellule, il che riduce fortemente la possibilità di contaminazione da mRNA con componenti batteriche. Ciò rende più facile produrre mRNA rispetto a pDNA in condizioni di buone pratiche di fabbricazione [12].
Infine, l’immunogenicità indotta da vettori può essere evitata per terapie con mRNA, a differenza di vettori virali o particelle simili a virus, che possono suscitare una risposta immunitaria specifica contro le proteine virali esposte [13]. Le applicazioni terapeutiche specifiche dell’mRNA, attualmente in fase di studio, includono
- i) vaccinazione contro il cancro e le malattie infettive,
- (ii) terapia sostitutiva delle proteine, e
- (iii) modifica genetica.
- La tabella 1 riassume esempi di studi clinici in corso su candidati vaccini terapeutici e profilattici basati su mRNA.
Tabella 1 – Esempi di studi clinici in corso di vaccini a base di mRNA.
| mRNA | Meccanismo di azione | Malattia / condizione | Itinerario di amministrazione | Fase di studio | Sponsor / Collaboratore | Identificatore nazionale della sperimentazione clinica |
|---|---|---|---|---|---|---|
| MRNA terapeutico | ||||||
| Vaccino W_ova1 | Induzione di una risposta immunitaria antitumorale | Cancro ovarico | Endovenoso | Fase I | University Medical Center Groningen / BioNTech | NCT04163094 |
| Cellule di Langerhans (LC) elettroporate con mRNA CT7, MAGE-A3 e WT1 | Elettroporazione di cellule dendritiche con mRNA dell’antigene | Mieloma multiplo | Sottocutaneo | Fase I | Memorial Sloan Kettering Cancer Center | NCT01995708 |
| MRNA cellulare personalizzato | Immunizzazione con DC pulsate con antigeni tumorali codificati da mRNA | Cancro al cervello / Neoplasia Metastasi | Non specificato | Fase I | Guangdong 999 Brain Hospital / Pechino, Tricision, Trinomab, Jinan University Guangzhou | NCT02808416 |
| MRNA personalizzato | Immunizzazione con DC pulsate con mRNA personalizzato | Glioblastoma | Non specificato | Fase I | Guangdong 999 Brain Hospital / Beijing Tricision, Trinomab, Jinan University Guangzhou | NCT02808364 |
| MiHA mRNA | Immunizzazione con DC caricate con MiHA mRNA | Neoplasie ematologiche | Endovenoso | Fase I Fase II | Radboud University / ZonMw: The Netherlands Organization for Health Research and Development Dutch Cancer Society | NCT02528682 |
| WT1-mRNA | Immunizzazione con DC elettroporate con WT1-mRNA | Leucemia mieloide acuta | Non specificato | Fase II | Fondazione Zwi Berneman / Kom Op Tegen Kanker contro la Fondazione per la ricerca sul cancro – Fiandre (FWO: Fondazione per la ricerca) | NCT01686334 |
| CMV umano pp65-LAMP mRNA | Immunizzazione con DC pulsate con mRNA di CMV pp65-LAMP | Glioblastoma | Intradermico | Fase II | Gary Archer Ph.D./Duke University | NCT03927222 |
| MRNA personalizzato | Vaccino tumorale personalizzato con mRNA che codifica per neoantigene | Carcinoma squamoso esofageo avanzato, adenocarcinoma gastrico, adenocarcinoma pancreatico, adenocarcinoma colorettale | Sottocutaneo | Iscrizione | Changhai Hospital / Stemirna Therapeutics | NCT03468244 |
| mRNA [BI 1361849 (ex CV9202)] | Non specificato | Carcinoma polmonare metastatico non a piccole cellule | Non specificato | Fase I Fase II | Ludwig Institute for Cancer Research / Cancer Research Institute, New York City, Boehringer Ingelheim MedImmune, CureVac, PharmaJet | NCT03164772 |
| mRNA-5671 / V941 | Non specificato | Neoplasie, carcinoma, polmone non a piccole cellule, neoplasie pancreatiche, neoplasie colorettali | Intramuscolare | Fase I | Merck Sharp & Dohme | NCT03948763 |
| mRNA-4157 | Immunostimolanti | Tumori solidi | Non specificato | Fase I | Moderna / Merck Sharp e Dohme | NCT03313778 |
| mRNA-4157 | Immunoterapia con il vaccino antitumorale personalizzato | Melanoma cutaneo | Non specificato | Fase II | Moderna / Merck Sharp e Dohme | NCT03897881 |
| MRNA personalizzato | Codifica neoantigene | Cancro esofageo Cancro polmonare non a piccole cellule | Sottocutaneo | Iscrizione | Stemirna Therapeutics / Il primo ospedale affiliato dell’Università di Zhengzhou | NCT03908671 |
| mRNA-3704 | Stimolanti alfa-galattosidasi; Stimolanti metilmalonil CoA mutasi; Stimolanti della sintesi proteica | Acidemia metilmalonica, metabolismo, errori congeniti | Endovenoso | Fase I Fase II | Moderna | NCT03810690 |
| mRNA-2416 | Modulatori ligando OX40 | Tumori solidi o linfomi recidivanti / refrattari | Intratumorale | Fase I | Moderna | NCT03323398 |
| mRNA-2752 | Stimolanti della proteina IL36G; Stimolanti dell’interleuchina 23; Modulatori ligando OX40 | Tumori solidi o linfomi recidivanti / refrattari | Intratumorale | Fase I | Moderno / AstraZeneca | NCT03739931 |
| MRNA profilattico | ||||||
| mRNA-1647, mRNA-1443 | Non specificato | Citomegalovirus | Non specificato | Fase I | Moderna | NCT03382405 |
| mRNA-1893 | Non specificato | Virus Zika | Non specificato | Fase I | Moderna / Biomedical Advanced Research and Development Authority | NCT04064905 |
| mRNA-1653 | Un vaccino combinato per metapneumovirus umano e virus della parainfluenza umana di tipo 3 | Metapneumovirus umano e virus parainfluenzale umano | Non specificato | Fase I | Moderna. | NCT03392389 NCT04144348 |
| mRNA-1944 | Codifica per un anticorpo monoclonale anti-virus Chikungunya | Virus Chikungunya | Parenterale | Fase I | Moderna | NCT03829384 |
| mRNA-1653 | Immunostimolanti | Metapneumovirus e virus parainfluenzale | Parenterale | Fase I | Moderna | NCT03392389 |
| CV7202 | Immunostimolanti | Rabbia | Intramuscolare | Fase I | CureVac | NCT03713086 |
Due classi di mRNA, ovvero mRNA non replicante e autoamplificante, sono comunemente usate come vettori vaccinali. L’mRNA non replicante codifica solo per l’antigene proteico di interesse, mentre l’mRNA autoamplificante codifica anche per le proteine che consentono la replicazione dell’RNA [14].
I vaccini basati sull’mRNA autoamplificante codificano il genoma dell’RNA di un virus a RNA a filamento singolo, ad esempio un alfavirus, un flavivirus [15] o un picornavirus [7].
Sono progettati per aumentare la durata e il livello di espressione, così come la successiva risposta immunitaria indotta dagli antigeni codificati. Amplificano in modo efficiente la produzione di mRNA sub-genomico che codifica gli antigeni di interesse dopo un singolo ciclo di replicazione. Mentre sia l’mRNA autoamplificante che l’mRNA non replicante trovano applicazione nei vaccini profilattici per le malattie infettive, l’mRNA non replicante viene utilizzato per i vaccini contro il cancro
Farmacologia fondamentale dei vaccini a mRNA
L’mRNA trascritto in vitro (IVT) è impiegato terapeuticamente poiché imita l’mRNA nativo completamente maturo presente nel citosol eucariotico [16]. Ciò può essere ottenuto mediante trasfezione ex vivo di cellule con mRNA che vengono poi trasferite in modo adottivo o mediante consegna diretta in vivo dell’mRNA IVT al citosol [17].
Questi approcci sono esplorati per l’ingegneria del genoma, la riprogrammazione genetica, le cellule T adottive e le immunoterapie per malattie infettive basate su cellule T adottive e dendritiche (DC), regimi di tolleranza per il trattamento delle allergie e terapie sostitutive delle proteine.
Sia la trasfezione ex vivo che la trasfezione diretta in vivo consentono alle cellule bersaglio di sintetizzare le proteine codificate in situ, dove l’mRNA viene utilizzato come modello e la proteina (le proteine) rappresenta il prodotto attivo. L’open reading frame (ORF) dell’mRNA maturo che codifica per le proteine di interesse (il prodotto attivo) contrassegnato rispettivamente dai codoni di inizio e fine è affiancato da regioni non tradotte (UTR) e idealmente è costituito da un limite di 5 ‘e una coda poli (A) [3].
L’attività farmacodinamica dell’mRNA sia nativo che IVT si svolge nel citosol (Figura 1).
Tuttavia, a differenza dell’mRNA endogeno, che viene trascritto dal DNA nel nucleo ed entra nel citosol attraverso l’esportazione nucleare, l’mRNA IVT entra nel citosol da una fonte extracellulare [18].
Una volta che l’mRNA IVT viene consegnato al citosol, la sua farmacologia è governata dagli stessi complessi meccanismi cellulari che regolano la stabilità e la traduzione dell’mRNA endogeno. L’mRNA IVT ingegnerizzato assomiglia così strettamente all’mRNA endogeno che il meccanismo di traduzione cellulare viene utilizzato senza soluzione di continuità per sintetizzare una proteina che può subire modifiche post-traduzionali, risultando infine in prodotti proteici maturi.
Nel caso dei vaccini , questo prodotto proteico maturo rappresenta l’antigene, che può suscitare potenti risposte umorali e cellulo-mediate potenti patogene-specifiche. Tuttavia, la destinazione intracellulare finale è determinata dalla o dalle sequenze naturali o ingegnerizzate del peptide segnale o dal dominio transmembrana [7].
Pertanto, i vaccini a mRNA possono essere progettati per il rilascio delle proteine codificate al compartimento cellulare desiderato per una presentazione e / o funzione adeguate [19].

Meccanismo d’azione dei vaccini a mRNA. 1. L’mRNA viene trascritto in vitro (IVT) da uno stampo di DNA in un sistema privo di cellule. 2. L’mRNA IVT viene successivamente trasfettato in cellule dendritiche (DC) tramite (3) endocitosi. 4. L’mRNA intrappolato subisce una fuga endosomiale e viene rilasciato nel citosol. 5. Usando il meccanismo di traduzione delle cellule ospiti (ribosomi), l’mRNA viene tradotto in proteine antigeniche. La proteina antigenica tradotta subisce una modifica post-traduzionale e può agire nella cellula in cui viene generata. 6. In alternativa, la proteina viene secreta dalla cellula ospite. 7. La proteina dell’antigene è degradata dal proteasoma nel citoplasma. Gli epitopi peptidici antigenici generati vengono trasportati nel reticolo endoplasmatico e caricati su molecole di classe I del complesso principale di istocompatibilità (MHC) (MHC I). 8.+ Risposte delle cellule T dopo riconoscimento del recettore delle cellule T e co-stimolazione appropriata. 9. Le proteine esogene vengono assorbite dalle DC. 10. Sono degradati negli endosomi e presentati tramite la via MHC II. Inoltre, per ottenere l’aiuto dei linfociti T affini nelle cellule che presentano l’antigene, la proteina dovrebbe essere instradata attraverso la via MHC II. 11. Gli epitopi peptidici antigenici generati vengono successivamente caricati su molecole MHC II. 12. I complessi epitopici MHC II-peptide caricati sono presentati sulla superficie delle cellule, portando all’induzione delle risposte delle cellule T CD4 + antigene-specifiche . Gli antigeni esogeni possono anche essere elaborati e caricati su molecole MHC di classe I tramite un meccanismo noto come presentazione incrociata (non mostrato in figura). La figura è stata creata con BioRender.com.
La farmacocinetica dell’mRNA IVT è determinata dall’emivita dell’mRNA e dalla proteina matura risultante dopo la modifica post-traduzionale. I due principali fattori che influenzano la biodisponibilità dell’mRNA esogeno nel citosol sono
- (i) rapida degradazione mediata da RNasi e
- (ii) mancanza di diffusione passiva attraverso la membrana plasmatica a causa dell’alto peso molecolare e repulsione elettrostatica tra le cariche negative della membrana cellulare rivestita di proteoglicano e le molecole di mRNA caricate negativamente [20].
L’mRNA nudo viene rapidamente degradato dalle RNasi extracellulari, ostacolando così la sua consegna efficiente ed efficacia. È stato dimostrato che un’ampia gamma di reagenti di trasfezione in vitro e in vivo protegge l’mRNA dalla degradazione e facilita l’assorbimento cellulare e la fuoriuscita endosomiale dell’mRNA.
Grandi sforzi sono stati dedicati al miglioramento della stabilità enzimatica dell’RNA, come discusso più avanti [3]. In definitiva, l’mRNA IVT composto da nucleotidi naturali viene metabolizzato da meccanismi fisiologici intrinseci, riducendo così il rischio di tossicità indotta dai metaboliti.
Pertanto, i problemi di consegna possono essere superati mediante approcci che includono l’incapsulamento dell’mRNA in sistemi di rilascio di farmaci costituiti da molecole cationiche, lipidi, polimeri e nanoparticelle [21], nonché bersagli DC [22].
Inoltre, è stato dimostrato che metodi di trasfezione fisica come l’elettroporazione aumentano l’efficienza di consegna di mRNA grandi e auto-amplificati in vivo, misurando l’espressione genica reporter e l’immunogenicità dei geni che codificano le proteine dell’involucro dell’HIV [23].
Approcci per migliorare la stabilità dell’mRNA
Lo sviluppo di farmaci a base di mRNA risale al 1990 con l’espressione con successo di un numero di proteine differenti dopo l’iniezione di mRNA che codifica queste proteine direttamente nei muscoli dei topi [24].
Questo ha portato a
- (i) la sperimentazione del primo vaccino a base di mRNA nel 1993, che ha dimostrato di indurre una risposta dei linfociti T citotossici anti-influenzali nei topi [25], e
- (ii) la prima vaccinazione con antigeni tumorali codificanti mRNA nel 1995 [26].
Queste dimostrazioni inceptive hanno ratificato il potenziale dell’mRNA per
- (i) espressione in situ di proteine specifiche e
- (ii) per l’induzione dell’immunità cellulare e umorale antigene-specifica protettiva.
Tuttavia, il campo è stato trascurato per quasi dieci anni fino a quando è stato scoperto il potenziale dell’applicazione in vivo dell’mRNA, cioè l’induzione di specifici linfociti T citotossici e di anticorpi [27].
I progressi nel campo dell’mRNA sono stati lenti a causa della natura labile dell’mRNA, il che rende gli esperimenti che impiegano mRNA non modificato molto impegnativi a meno che le precauzioni per manipolare l’mRNA non siano strettamente rispettate [28]. L’attenzione è stata invece rivolta ai farmaci a base di DNA, poiché il DNA è più stabile dell’RNA.
In un sistema cell-free, l’mRNA può essere sintetizzato mediante IVT di uno stampo di DNA (ad esempio, un plasmide linearizzato o un prodotto PCR), che codifica tutti gli elementi strutturali di un mRNA funzionale. Per eseguire una reazione IVT, sono necessari tutti gli elementi del processo di trascrizione naturale, vale a dire, uno stampo di DNA, una RNA polimerasi e blocchi di nucleotidi.
Durante la successiva purificazione dell’mRNA, il DNA stampo viene spesso degradato mediante l’aggiunta di DNasi, seguita dalla purificazione mediante altri metodi convenzionali per l’isolamento dell’mRNA, ad esempio precipitazione e cromatografia. Questo processo si traduce in prodotti mRNA altamente puri pronti per l’uso [29,30,31]. Diverse strategie sono state perseguite per far fronte alla mancanza intrinseca di stabilità e potenziale immunogenicità dell’mRNA, che sono discusse più avanti.
Stabilizzazione molecolare
Le strategie che includono l’ingegnerizzazione di sequenze e / o struttura per migliorare la stabilità dell’mRNA (estendere l’emivita) e la traduzione sono spesso fondamentali per aumentare i livelli di espressione della proteina. Le tecniche impiegate per ottenere ciò includono l’allungamento della coda poli (A), la modifica del cappuccio 5 ‘, l’ingegneria degli UTR e dei modelli di sequenza nell’ORF e / o l’incorporazione di nucleotidi modificati (Figura 2).

Struttura dell’mRNA trascritto in vitro (IVT) e strategie di modifica comunemente utilizzate. Il design dell’mRNA IVT si basa sul modello dell’mRNA eucariotico e consiste in regioni non tradotte (UTR) da 5 ‘cap, 5’ e 3 ‘, un frame di lettura aperto (ORF) che codifica antigeni e un 3 ‘poli (A) coda. L’mRNA dell’IVT può essere modificato in uno o più siti, ad esempio modificando le calotte, gli UTR e / o la coda poli (A), per modulare la durata e il profilo cinetico dell’espressione proteica. eIF4E, fattore di inizio della traduzione eucariotica 4E.
Cap Analog
Un analogo del cappuccio sintetico può essere facilmente aggiunto all’mRNA perché il cappuccio terminale 5 ‘non è codificato dal modello di DNA. L’mRNA eucariotico naturale ha un cappuccio di 7-metilguanosina (m7G) accoppiato all’mRNA durante il processo di trascrizione tramite un ponte 5′-5’-trifosfato (ppp) [32].
La struttura m7GpppN all’estremità 5 ‘del cappuccio dell’mRNA svolge diverse funzioni [33]. In primo luogo, protegge l’mRNA dalla rapida degradazione da parte delle esonucleasi. In secondo luogo, svolge un ruolo indispensabile durante la traduzione perché il fattore di iniziazione eucariotico (eIF) 4E riconosce e si lega al cappuccio dell’mRNA.
Svolge inoltre un ruolo nell’impedire ai sensori immunitari innati di riconoscere l’mRNA [34]. L’mRNA può contenere uno dei tre cappucci distinti, cioè, cap-0 [m7G (5 ‘) pppN1pN2p], cap-1 [m7G (5’) pppN1mpNp] e cap-2 [m7G (5 ‘) pppN1mpN2mp], rispettivamente [ 11].
Il capping dell’mRNA IVT può essere eseguito utilizzando due diversi approcci: Il primo approccio include l’aggiunta di una seconda fase con enzimi di capping ricombinanti derivati dal virus vaccinia dopo la trascrizione, risultando in un limite identico alla struttura del cappuccio eucariotico endogeno più frequente, ovvero 7 -metilguanosina (m7G) tappo [30].
In alternativa, un analogo del cappuccio sintetico può essere aggiunto durante la reazione di trascrizione in vitro, quindi il capping e la trascrizione in vitro vengono eseguiti in un unico passaggio. Questo approccio è denominato capping co-trascrizionale [35]. Uno dei principali svantaggi di questo approccio è la competizione tra l’analogo del cappuccio e il nucleotide GTP richiesto per la trascrizione in vitro, che alla fine si traduce in una frazione di mRNA non chiuso e tradizionalmente inattivo [36].
La frazione di mRNA uncapped contenente gruppi 5 ‘ppp è più immunostimolante, che può essere rettificata mediante trattamento con fosfatasi per rimuovere il gruppo ppp all’estremità 5’ dell’mRNA uncapped [37].
Vengono utilizzate tre diverse classi di analoghi del cap m7GpppG [38]:
- (i) analoghi anti-reverse cap (ARCA) [39],
- (ii) 3′-O-Me-m7GpppG [40], e
- (iii) ARCA modificati [41].
La ricerca iniziale sull’mRNA è stata eseguita utilizzando l’mRNA contenente l’analogo del cap m7 (GpppG) [42], ed è attualmente il cap dell’mRNA più comunemente usato negli studi clinici. Sfortunatamente, una frazione dell’analogo del cappuccio m7GpppG utilizzato durante la trascrizione in vitro viene incorporata nell’orientamento opposto e quindi non viene riconosciuta dai ribosomi, con conseguente minore attività traslazionale.
Per evitare ciò, è stato introdotto il cosiddetto ARCA con un solo gruppo 3′-OH invece di due gruppi 3′-OH (ARCA; m27,3′-OGpppG) per prevenire l’incorporazione nell’orientamento opposto [43]. È stato dimostrato che gli ARCA che giustappongono i tradizionali analoghi del cappuccio mostrano un’efficienza di trascrizione dell’RNA più di quattro volte superiore [44].
Inoltre, è stato riscontrato che la durata ed i livelli di espressione proteica sono aumentati nelle cellule trasfettate con RNA IVT con cappuccio ARCA [41]. Recentemente, sono stati introdotti nuovi tipi di analoghi del cappuccio modificati chimicamente, ad esempio, fosforotioato, fosforotiolato [35], imidifosfato [45], acido nucleico bloccato [46], legami boranofosfato [47] e altri tipi di modifiche, che forniscono il mRNA con resistenza al decapping da parte dell’enzima 2 di decapping dell’mRNA, che alla fine si traduce in una più lunga emivita dell’mRNA [48].
Convenzionalmente, l’RNA sintetico 5′-Cap 0-capped viene trascritto eseguendo una trascrizione in vitro, dove più dell’80% del GTP aggiunto alla reazione viene sostituito con un analogo del cappuccio dinucleotidico (cioè, m7G [5 ‘] ppp [ 5 ‘] G), con conseguente inizio della trascrizione con l’analogo del tappo [49].
Tuttavia, è stato dimostrato che questo approccio presenta una serie di inconvenienti legati all’efficienza, che sono stati superati dall’introduzione di ScriptCap ™. ScriptCap ™ prevede l’aggiunta di strutture cap-0 costruite enzimaticamente sui trascritti di RNA mediante l’impiego di un enzima di capping con efficienza di reazione del 100% [50]. L’mRNA eucariotico nativo può contenere un cap-1 o un cap-2 ma mai un cap-0 [11].
Pertanto, l’mRNA IVT dovrebbe contenere un cap-1 o un cap-2 per essere meno immunostimolante [51]. Un segno distintivo di queste strutture a cappuccio è lo stato di metilazione della posizione 2 ‘del 5’ penultimo e terzultimo nucleoside. Prima dell’avvento della nuova tecnologia CleanCap ™ introdotta da TriLink, l’mRNA sintetico con un cap-1 poteva essere preparato solo mediante capping enzimatico [52,53].
Tuttavia, cap-1 o cap-2 possono ora essere incorporati durante il capping co-trascrizionale con un’efficienza di capping di circa il 94% [52]. In particolare, è stato dimostrato che l’efficienza del capping è significativamente superiore all’efficienza raggiunta dal capping co-trascrizionale tradizionale con cap-0 o ARCA [54].
Regioni 5 ‘e 3’ non tradotte (UTR)
L’importanza dell’incorporazione di 5′- e 3′-UTR è stata notata durante la regolazione post-trascrizionale in vitro dell’espressione genica [55]. I numerosi ruoli che svolgono gli UTR includono
- (i) regolazione dell’esportazione di mRNA dal nucleo,
- (ii) regolazione dell’efficienza della traduzione [56],
- (iii) orchestrazione della localizzazione subcellulare [57], e
- (iv) stabilità dell’mRNA [58].
L’introduzione degli UTR finali dell’α-globina 3 ‘porta alla stabilizzazione dell’mRNA, mentre l’incorporazione degli UTR dell’estremità 5’ e 3 ‘della beta-globina porta a una maggiore efficienza traslazionale [59]. Il risultato ottimale si ottiene utilizzando due β-globina 3’-UTR allineate in una configurazione testa a coda.
Le UTR di α-globina e β-globina sono state incorporate per modificare l’RNA per una trascrizione in vitro ottimizzata seguita dall’elettroporazione dell’mRNA di cellule T autologhe [60] e dall’iniezione intranodale di RNA codificante l’antigene nudo [61].
Inoltre, le DC trasfettate con mRNA ottimizzato per UTR codificante per l’antigene sono state utilizzate in uno studio che ha coinvolto l’immunizzazione di individui sieropositivi al citomegalovirus e pazienti affetti da cancro [62]. In alcune situazioni, destabilizzare l’mRNA potrebbe essere un approccio praticabile per ridurre la durata della sintesi proteica. Ciò può essere ottenuto introducendo elementi ricchi di adenilato-uridilato nelle 3′-UTR dell’mRNA, con conseguente degradazione più rapida dell’mRNA e riduzione della durata dell’espressione della proteina [63].
Poly (A) Coda
La coda poli (A) gioca un ruolo significativo nella traduzione dell’mRNA e per la stabilità enzimatica dell’mRNA. La coda poli (A) si lega a diverse proteine leganti il poliadenosile (PABP) mentre lavora sinergicamente con sequenze 5’m7Gcap per regolare l’efficienza traslazionale [64].
Il fattore di inizio della traduzione eucariotica eIF4E si lega al tappo 5’m7G, che a sua volta si complessa con eIF4G ed eIF4A. La PABP interagisce quindi con il terminale N del fattore di inizio della traduzione eucariotica eIF4G, che forma un mRNP (ribonucleoproteina messaggera) o un complesso polisomico [65].
Il primo raffigura il complesso mRNA-proteina non ancora coinvolto nella sintesi proteica, mentre il secondo è già in fase di traduzione. Una coda di poli (A) adeguatamente lunga è necessaria per circolarizzare l’mRNA attraverso il legame dei PABP alla coda di poli (A) e al cappuccio [55,66]. È stato osservato che l’aumento della lunghezza della coda del poli (A) migliora l’efficienza della generazione di polisomi e di conseguenza influenza i livelli di espressione della proteina [67].
È stato dimostrato che un aumento graduale della lunghezza della coda poli (A) dell’mRNA IVT a 120 basi aumenta in modo proporzionale il livello di espressione della proteina, mentre un aumento del numero di basi oltre 120 non migliora ulteriormente l’espressione della proteina [68]. Le code di poli (A) possono essere aggiunte all’mRNA codificando la coda di poli (A) nello stampo di DNA o per estensione dell’RNA IVT dopo la trascrizione utilizzando la poli (A) polimerasi ricombinante.
Tuttavia, la poliadenilazione con poli (A) polimerasi ricombinante si traduce in una lunghezza della coda poli (A) variabile, producendo in tal modo mRNA poliadenilato con lunghezze variabili. Pertanto, l’approccio preferito è la generazione di code di poli (A) con lunghezza ben definita dagli mRNA trascritti da modelli di DNA codificanti la coda di poli (A) [69].
Le interazioni fisiche tra le estremità 5 ‘e 3’ dell’mRNA avvengono tra il cappuccio e la coda del poli (A) [70]. La coda di poli (A) svolge anche un ruolo nel prevenire il decapping e la degradazione dell’mRNA poiché la rimozione o l’accorciamento della coda di poli (A) a meno di 12 residui provoca la degradazione dell’mRNA attraverso la scissione della struttura del cappuccio da 5 ‘e da 5’ a 3 ‘digestione esonucleolitica o degradazione da 3’ a 5 ‘[71].
Strategie di formulazione
Nonostante il potenziale promettente dei vaccini a base di mRNA, il rilascio intracellulare efficiente di mRNA al citosol continua a rappresentare un ostacolo importante, specialmente per l’mRNA somministrato per via sistemica.
Il grande peso molecolare (105-106 Da) [21] e l’elevata densità di carica negativa dell’mRNA compromettono la permeazione dell’mRNA attraverso le membrane cellulari. È noto che l’assorbimento dell’mRNA in assenza di un sistema di rilascio è estremamente basso e l’emivita dell’mRNA è di circa 7 ore [72].
Inoltre, l’mRNA è una molecola intrinsecamente instabile, che è altamente soggetta a degradazione da parte di esonucleasi 5 ‘, esonucleasi 3’ ed endonucleasi [73]. Di conseguenza, i sistemi di rilascio sono indispensabili per il rilascio intracellulare dell’mRNA al sito d’azione terapeutico in vitro e in vivo [8,74].
Sono state studiate diverse strategie per migliorare la somministrazione di RNA, comprese strategie di iniezione migliorate come microiniezioni [75], patch di RNA [76], somministrazione basata su pistole geniche [77], condensazione della protamina [78], adiuvanti dell’RNA [79] e incapsulamento di RNA in nanoparticelle costituite da lipidi e / o polimeri [80].
Generalmente, l’mRNA IVT per il rilascio citosolico è formulato con un sistema di rilascio miscelato con un agente complessante [81], che può proteggere l’mRNA dalla rapida degradazione e facilitare l’assorbimento cellulare.
Sebbene il dogma generale nel campo sia che sono necessari vettori efficienti per migliorare sostanzialmente la trasfezione in vivo dell’mRNA, l’mRNA nudo è stato applicato in molti studi in vivo. Quindi, la sezione seguente discute la consegna di mRNA nudo, seguita da sezioni che discutono la consegna di mRNA basato su vettori [82,83,84,85].
RNA nudo
La strategia di somministrazione più semplice comprende l’iniezione intramuscolare (im) di mRNA nudo e la prova del concetto è stata originariamente dimostrata dall’espressione del gene reporter in vivo nei topi [24]. Da allora, l’efficacia dell’mRNA nudo è stata confermata con iniezioni im [86], sottocutanee (sc) [87] o intradermiche (id) [88]. La somministrazione di id e sc di mRNA ha dimostrato di mediare la guarigione di varie malattie della pelle e di migliorare la guarigione delle ferite mediante l’espressione in situ di proteine specifiche nella pelle [75,89].
L’adozione di questo approccio aggira diversi ostacoli altrimenti associati alla somministrazione sistemica di mRNA, ad esempio, l’eliminazione dal flusso sanguigno attraverso il fegato, i reni e la milza [75]. È stata dimostrata una traduzione molto efficiente della proteina codificata per mRNA somministrato sc [90,91,92].
È interessante notare che a volte è stata misurata una traduzione più efficiente rispetto ai sistemi di rilascio basati su nanoparticelle caricate con mRNA [90,93]. Quindi, elude la necessità di impiegare vettori, contribuendo infine a ridurre i costi e il rischio potenziale. Un altro vantaggio della somministrazione di vaccini a base di mRNA per via SC è che le risposte immunitarie sia cellulari che umorali sono indotte [94] perché l’mRNA è espresso sia dalle DC residenti nella pelle [95] che dalle cellule non immunitarie [96].
Tuttavia, lo strato corneo più esterno dell’epidermide funge da barriera contro l’assorbimento dei farmaci somministrati per via topica [97]. Finora, sono stati adottati vari approcci per superare questa barriera, inclusi quelli fisici (p. Es., Microporazione [98], microaghi [99] e iniezione a getto [100,101]), attivi (p. Es., Elettroporazione [102], ionoforesi [103] e sonoforesi [104]) e metodi passivi (es. nanoparticelle [105] e liposomi [106]).
L’elettroporazione e la sonoporazione possono permeabilizzare transitoriamente la pelle per mezzo di impulsi elettrici e ultrasuoni a bassa frequenza, rispettivamente, per il trasporto efficiente dei geni nella pelle [107]. Il fattore di crescita endoteliale vascolare codificante mRNA modificato-A è stato formulato in soluzione salina tamponata con citrato senza l’uso di un sistema di rilascio ed è stato utilizzato per la vaccinazione id di pazienti con diabete di tipo 2 [108].
Tuttavia, nonostante la vasodilatazione pronunciata, prolungata, dose-dipendente e specifica del carico, l’aumento del flusso sanguigno, la sovraregolazione del metabolismo dell’ossigeno, l’angiogenesi e la formazione di neovasi nei modelli animali, l’attività vasodilatatoria e angiogenica non si è tradotta nell’uomo [108,109].
L’erogazione basata su microneedle è anche una tecnica efficiente in cui vengono impiegati patch / array di aghi delle dimensioni di un micron composti da eccipienti polimerici o zuccheri solubili in acqua, in cui è incorporato l’mRNA [76]. I cerotti / matrici forniscono la resistenza meccanica necessaria affinché l’ago permea lo strato corneo e penetri negli strati vitali della pelle.
Dopo l’iniezione, a seconda del tipo di microaghi, i cerotti / array si degradano / si dissolvono e il farmaco incapsulato viene rilasciato nel fluido interstiziale della pelle [75]. La somministrazione di mRNA utilizzando microaghi solubili fornisce un vantaggio importante, ovvero la somministrazione in una forma di dosaggio solida elude la necessità di trattare l’mRNA in una forma di dosaggio liquida [110], che quindi elimina la minaccia proveniente dalla contaminazione da RNasi, insieme all’aumento dell’mRNA stabilità e durata di conservazione [76].
È stato dimostrato che l’endocitosi mediata dal recettore Scavenger e la micropinocitosi sono i meccanismi di assorbimento attivo dell’mRNA nudo nelle DC immature [16]. Tuttavia, l’mRNA nudo mostra una breve emivita plasmatica, è soggetto alla degradazione della ribonucleasi e incontra difficoltà nell’entrare nella cellula. Pertanto, sono stati proposti sistemi di rilascio per proteggere l’mRNA e schermare la sua carica negativa.
Vettori virali
La consegna dell’mRNA può essere mediata da vettori virali e non virali. I vettori non virali possono essere ulteriormente classificati in sistemi di rilascio basati su lipidi, sistemi di rilascio basati su polimeri e sistemi ibridi polimero-lipide [111]. Per il rilascio dell’RNA virale, c’è stato un grande interesse nell’ingegnerizzazione di virus adeno-associati per trasportare carichi di acidi nucleici [112].
I virus geneticamente modificati sono solitamente impiegati per la consegna di mRNA / gene. I geni di questi virus sono parzialmente o completamente sostituiti con geni modello o terapeutici. Un vantaggio dei virus a RNA è che vengono replicati ed espressi localmente nel citoplasma.
I virus a RNA a filamento positivo si distinguono per una sequenza genomica che può essere tradotta direttamente in proteine di interesse dai ribosomi ospiti. In particolare, per il rilascio di mRNA sono stati impiegati alfavirus (ad esempio, Sindbis e Semliki Forest virus) [113], picornavirus [114] e flavivirus [115] (ad esempio, virus Kunjin). Vari vettori alfavirus possono essere usati per esprimere alti livelli di proteine esogene in un ampio spettro di ospiti [116].
Gli approcci comunemente usati includono la sostituzione diretta di geni strutturali con espressione eterologa o il posizionamento dei geni non strutturali a valle del promotore subgenomico dell’RNA [117]. Tuttavia, gli alfavirus inducono gravi effetti citopatogeni, che limitano la loro applicazione nella terapia genica, sebbene possano essere impiegate strategie differenti per superare questa sfida [118].
Alcune di queste strategie includono l’ingegnerizzazione di vettori mutanti con citotossicità mitigata e inducibilità alla temperatura, e vettori autoinattivanti con mutazioni puntiformi nel gene nsP2 (specialmente in posizione 726, 259 e 650) [119]. Il virus Sendai (SeV) (virus parainfluenzale murino di tipo 1 o virus emoagglutinante del Giappone) che appartiene alla famiglia Paramyxoviridae è degno di nota per la sua popolare applicazione come vettore.
È favorito per i suoi livelli elevati ma transitori di espressione genica, ampia specificità della cellula ospite, bassa patogenicità e forte immunogenicità [120]. In quanto piattaforma vaccinale, il virus dell’encefalite venezuelana è di particolare interesse [121]. Questi vaccini comprendono il vaccino virale vivo attenuato TC-83 e una sua varietà inattivata con formalina è denominata C-84, che aumenta l’efficacia e aumenta la durata dell’immunità dopo la somministrazione del vaccino TC-83 attraverso diverse vie di somministrazione, intranasale è il più utilizzato [122].
Tuttavia, l’uso di vettori virali incorpora inconvenienti cruciali associati all’integrazione del genoma e al possibile rigetto dell’ospite (immunogenicità e citotossicità) tra gli altri [123], provocando quindi la necessità di vettori non virali per la consegna di mRNA [10].
Vettori a base di polimeri
Il destrano dietilamminoetile (DEAE) è stato il primo polimero ad essere testato come reagente di rilascio per l’mRNA IVT [124]. Successivamente, è stato dimostrato che la trasfezione dell’mRNA mediata dai lipidi è da 100 a 1000 volte più efficiente del DEAE-destrano [125]. Questa scoperta ha bloccato il progresso dei trasportatori polimerici e ha aperto la strada ai reagenti di trasfezione a base di lipidi per gli acidi nucleici, compreso l’mRNA.
Uno studio completo ha confrontato i polimeri poli-beta-ammino-esteri (PBAE) e polietilenimina (PEI) con il reagente di trasfezione commerciale Lipofectamine ™ 2000 e l’1,2-dioleoil-3-trimetilammonio propano (DOTAP) / 1,2-dioleoil-sn -glicero-3-fosfoetanolamina (DOPE) per risposte funzionali, antigene-specifiche dei linfociti T dopo la somministrazione di mRNA [126].
Tutti i portatori sono stati complessati con mRNA che codifica per il bavaglio dell’antigene HIV-1. Le cellule T secernenti IFN-γ specifiche del bavaglio sono state misurate nella milza e nei linfonodi di topi immunizzati con mRNA del bavaglio complessato con lipidi cationici ma non nei topi immunizzati con mRNA nudo e complesso con polimero. Il PEI ei suoi derivati sono tra i polimeri cationici più comunemente impiegati [127].
Sono solubili in acqua, mostrano un’alta densità di carica positiva associata ai gruppi amminici e sono portatori di mRNA provati per la trasfezione in vitro [128]. Tuttavia, la PEI mostra problemi di tossicità a causa dell’elevato peso molecolare (> 25 kDa), che può derivare dall’adsorbimento di proteine sieriche anioniche sulla superficie polyplex tra polimeri cationici e proteine plasmatiche sieriche anioniche.
Tuttavia, l’aumento delle dimensioni risultante è solo transitorio poiché le proteine adsorbite sulla superficie dei poliplessi prevengono l’aggregazione particella-particella a lungo termine [129]. Sono stati compiuti vari sforzi per mitigare queste sfide. La prima prova di concetto per la trasfezione del vaccino mRNA sicura ed efficace utilizzando polimero cationico è stata ottenuta mediante somministrazione intranasale di 2 kDa PEI coniugato a ciclodestrina.
La ciclodestrina coniugata a PEI ha consentito la delocalizzazione della densità di carica sullo scheletro poliamminico, riducendo così la citotossicità e allo stesso tempo mantenendo i gruppi protonabili, con conseguente miglioramento della trasfezione [130]. Le nanoparticelle polimeriche composte da polimeri biodegradabili, ad esempio, poli (acido lattico-co-glicolico) (PLGA), sono adatte per l’incorporazione di molecole idrofobiche e caricate positivamente. Forniscono una buona stabilità colloidale, bassa tossicità e possibilità di rilascio prolungato. Tuttavia, a causa della natura anionica del PLGA a pH fisiologico [131], l’efficienza di incapsulamento dell’mRNA è molto bassa.
I portatori a base di polimeri mostrano un notevole potenziale per la terapia genica grazie alla sostanziale efficienza di trasfezione e alla tossicità tollerabile [132]. Una serie di copolimeri a blocchi multifunzionali, ovvero dimetilamminoetil metacrilato (DEAMA), poli (etilenglicole) metacrilato e DEAEMA-co-n-butil metacrilato, ha dimostrato un’efficienza di trasfezione del 77% e 50% nei macrofagi RAW 264,7 e DC2,4 cellule dendritiche, rispettivamente, mostrando così un potenziale come vettore per la somministrazione di vaccini intracellulari basati su mRNA [133].
Sebbene siano stati testati diversi tipi di polimeri e copolimeri, la correlazione tra la struttura dei polimeri e la loro risposta biologica, ad es. Trasfezione e tossicità, è risultata scarsa e, quindi, la progettazione di vari sistemi di rilascio basati su polimeri si basa piuttosto sull’empirico rispetto agli approcci razionali [134]. Nonostante i vantaggi sopra menzionati, i sistemi di rilascio basati su polimeri non sono clinicamente avanzati come i sistemi di rilascio a base di lipidi a causa della loro polidispersione e delle sfide relative al metabolismo di polimeri di grande peso molecolare [21].
Vettori a base di lipidi
I vettori basati su lipidi o composti simili ai lipidi (lipidoidi) rappresentano i portatori di geni non virali più comunemente usati [21]. Vari lipidi sintetici e di derivazione naturale sono stati impiegati per formare liposomi (i complessi di liposomi e acidi nucleici sono indicati come lipoplessi) o nanoparticelle lipidiche (LNP), entrambi i quali sono stati segnalati per fornire in modo efficiente vaccini a base di mRNA (Tabella 2) .
Gli LNP sono spesso formulati utilizzando lipidi cationici che mostrano ammine terziarie o quaternarie per incapsulare l’mRNA polianionico. I lipidi cationici incapsulano spontaneamente l’mRNA caricato negativamente, mediato da una combinazione di interazioni elettrostatiche attraenti con l’RNA e interazioni idrofobiche, e quindi sono stati usati da soli o in combinazione per la lipofection dell’mRNA.
Il primo utilizzo segnalato di LNP come sistema di rilascio per mRNA è avvenuto nel 2015, con il sistema di rilascio costituito da lipide cationico ionizzabile / fosfatidilcolina / colesterolo / PEG-lipide nel rapporto di (50: 10: 38,5: 1,5 mol / mol) [86 ]. Esempi di lipidi cationici includono, ad esempio, 1,2-di-O-ottadecenil-3-trimetilammonio propano (DOTMA) [135], DOTAP [136] e zwitterionico DOPE [137,138].
Sono strutturalmente indicati da un headgroup cationico, un gruppo coda idrofobo e un gruppo di collegamento in mezzo [139].
Tuttavia, è stato osservato che i lipidi cationici mostrano reazioni pro-infiammatorie ed effetti collaterali indesiderati [140].
Pertanto, i lipidi neutri sono anche incorporati nei liposomi cationici per diminuire la tossicità e raggiungere alti livelli di trasfezione in vivo [106].
Il meccanismo di rilascio dell’mRNA mediato da LNP non è completamente compreso, ma si suggerisce che gli LNP siano internalizzati dall’endocitosi e siano attaccati elettrostaticamente e fusi con la membrana cellulare tramite fasi lipidiche non a doppio strato invertite [21].
Tavolo 2
Esempi di sistemi di somministrazione di farmaci nanoparticellari per la consegna di mRNA.
| Sistemi di somministrazione di farmaci | Composizione | RNA | Malattia / condizione | Riferimenti |
|---|---|---|---|---|
| Polimeri | Poly (glycoamidoamine) | MRNA dell’eritropoietina (EPO) | Anemia e mielodisplasia | [168] |
| Polietilenimmina | HIV-1 gag mRNA | HIV | [169] | |
| Poli (β-amminoestere) (PBAE) | eGFP mRNA | N / A | [170] | |
| Copolimero Triblock (composto da DMAEMA, PEGMA, DEAEMA e BMA) | eGFP e ovoalbumina | N / A | [171] | |
| DEAE-Dextran | MRNA che codifica per la luciferasi | N / A | [172] | |
| Lipidi | DOTAP / DOPE | HxB-2 HIV-1 Antigene Gag mRNA | HIV | [126] |
| DOPE / DC-Colesterolo [2: 1] | eGFP mRNA | N / A | [106] | |
| DOTAP / Liposoma colesterolo [1: 1] con DSPE-PEG e DSPE-PEG-AA | HSV I Timidina chinasi mRNA | Cancro | [142] | |
| C12-200: Colesterolo: DOPE: C14-PEG2000 | EPO mRNA | N / A | [157] | |
| A18 | Ovoalbumina mRNA | Melanoma | [173] | |
| cKK-E12 | MRNA dell’anticorpo HER2 | Cancro | [174] | |
| (6Z, 9Z, 28Z, 31Z) -eptatriaconta-6,9,28,31-tetraen-19-il 4- (dimetilammino) butanoato (MC3), DSPC, colesterolo e 1,2-dimiristoil-rac-glicerolo, glicole metossipolietilenico (PEG2000-DMG) | Eritropoietina umana | [175] | ||
| DOTAP / DOPE [1: 1] | Antigene HIV-1 Gag mRNA | HIV | [126] | |
| 3β- [N- (N ‘, N’-dimetilamminoetano) carbamoile] (DC-colesterolo) / DOPE (1: 2) | eGFP mRNA | N / A | [106] | |
| NP ibride polimeriche lipidiche | TT3: DOPE: Colesterolo: DMG-PEG2000 con nucleo PLGA | Firefly luciferase (FLuc) mRNA e eGFP mRNA | N / A | [160] |
| PBAE: C14-PEG2000 | FLuc mRNA | N / A | [161] | |
| PBAE: EDOPC / DOPE / DSPE-PEG | Ovoalbumina mRNA | N / A | [176] | |
| PBAE: DOPC, DOTAP e DSPE-PEG | eGFP mRNA | N / A | [105] | |
| Peptidi e ibridi peptide-polimero | PepFect14 | eGFP mRNA | Cancro ovarico | [164] |
| RALA | eGFP mRNA OVA mRNA | N / A | [165] | |
| RALA-PLA | eGFPmRNA FLuc mRNA | N / A | [167] |
BMA: butil metacrilato; DEAE: dietilamminoetile; DEAEMA: dietilamminoetil metacrilato; DMAEMA: dimetilamminoetil acrilato; DOPE: dioleoilfosfatidiletanolammina; DOTAP: propano di dioleoil-3-trimetilammonio; DSPC: dipalmitoilfosfatidilcolina; DSPE-PEG: 1,2-distearoil-sn-glicero-3-fosfoetanolammina-N- [ammino (polietilenglicole); DSPE-PEG-AA: DSPE-PEG-anisammide; eGFP: proteina fluorescente verde potenziata; HER2: recettore 2 del fattore di crescita epidermico umano; HIV: virus dell’immunodeficienza umana; HSV: virus dell’herpes simplex; N / A: non applicabile; PEGMA: poli (glicole etilenico) metacrilato; PLGA: poli (acido lattico-co-glicolico); TT: N1, N3, N5-tris (2-amminoetil) benzene-1,3,5-tricarbossammide; PLA: acido polilattico.
I liposomi sono strutture a membrana chiusa, che si formano per autoassemblaggio quando i fosfolipidi sono dispersi in sistemi acquosi [141]. Sono costituiti da almeno un doppio strato fosfolipidico, che imita la struttura della membrana cellulare che racchiude un nucleo acquoso [8]. DOTAP / DOPE con un rapporto molare 1: 1 è stato segnalato come un efficace agente di trasfezione per mRNA che codifica per l’antigene HIV-1 Gag, che ha indotto con successo una risposta immunitaria antigene-specifica in vivo nei topi [126].
Inoltre, liposomi a base di 3β-N- (N ‘, N’-dimetilamminoetano) carbamoil-colesterolo) / DOPE in un rapporto [1: 2] hanno ottenuto un’elevata efficienza di incapsulamento dell’mRNA della proteina fluorescente verde (eGFP), insieme a un eGFP elevato espressione in vitro [106]. Inoltre, un ulteriore LNP multicomponente ha mostrato un effetto soppressore del tumore quando caricato con il virus herpes simplex I (HSV I) timidina chinasi codificante per mRNA.
Gli LNP erano composti da liposomi DOTAP / colesterolo [1: 1] insieme a 1,2-distearoil-fosfatidiletanolammina (DSPE) -polietilenglicole (PEG) e DSPE-PEG-anisamide (AA) [142]. Il principio alla base della loro efficacia può essere riassunto come una combinazione delle sue interazioni elettrostatiche attribuibili a cariche opposte e interazioni idrofobiche con mRNA.
Inoltre, le capacità di fuga endosomiale e le proprietà autoassemblanti che danno luogo a strati uniformi che racchiudono nuclei polimerici contribuiscono anche all’ampia applicazione di lipidi cationici [143]. Tuttavia, gli studi in vivo sono più impegnativi a causa della rapida eliminazione dei lipidi cationici da parte del sistema fagocitico mononucleare [144].
I lipidi cationici costituiti da un solo gruppo di testa di ammonio quaternario pongono problemi di sicurezza come tossicità e immunogenicità in vitro [145] e in vivo [146]. Ad esempio, i liposomi cationici quando somministrati per via endovenosa possono indurre epatotossicità [147] e possono innescare una forte risposta IFN-γ nei topi con conseguente infiammazione [148,149].
Inoltre, i lipidi caricati positivamente, ad esempio DOTAP e DOTMA, possono essere neutralizzati dalle proteine sieriche anioniche, determinando tossicità e ridotta efficacia [150]. Inoltre, possono sorgere sfide come il legame illimitato alle proteine, l’instabilità colloidale e la fuoriuscita di farmaci [151].
In alternativa, sono stati introdotti nuovi vettori di rilascio genico contenenti lipidi ionizzabili [152] e materiali simili ai lipidi chiamati lipidoidi [153] per superare le sfide poste dai lipidi cationici convenzionali pur mantenendo le loro proprietà di trasfezione vantaggiose.
I lipidi ionizzabili per la trasfezione dell’mRNA sono caricati positivamente a pH basso (che aiuta nella complessazione dell’mRNA quando viene eseguita in tampone acido) ma sono neutri a pH fisiologico (per una ridotta tossicità post-iniezione) [154]. A differenza dei lipidi cationici convenzionali, questi lipidoidi mostrano una serie di ammine secondarie e terziarie che consentono interazioni più efficienti con l’mRNA senza aumentare notevolmente la carica complessiva del sistema di rilascio [155].
L’incapsulamento dell’mRNA nelle nanoparticelle serve a proteggere fisicamente gli acidi nucleici dalla degradazione e, a seconda della chimica specifica, può aiutare nell’assorbimento cellulare e nella fuga endosomica [156]. La combinazione del lipide ionizzabile C12-200, colesterolo, DOPE e C14-PEG2000 con un rapporto molare di 3,5: 4,65: 1,6: 0,25, rispettivamente, incapsulando l’eritropoietina mRNA (EPO-mRNA) ha mostrato un’elevata efficacia in vivo quando iniettata nei topi, misurata come espressione cellulare di EPO [157]. L’enfasi sulla piattaforma di nanoparticelle per la consegna di mRNA è in parte dovuta all’applicazione di sistemi di rilascio di DNA e siRNA consolidati.
Nanoparticelle ibride polimero lipidico
È stato dimostrato in precedenza che le nanoparticelle ibride polimero-lipidico (LPN) mostrano un rilascio funzionale efficace di siRNA in vitro [158] e il rilascio terapeutico di siRNA in vitro e in vivo [159]. Questo sistema di rilascio ibrido ha anche mostrato risultati promettenti per la consegna di mRNA, con l’mRNA incapsulato in una nanoparticella ibrida composta dal materiale lipidico N1, N3, N5-tris (2-amminoetil) benzene-1,3,5- tricarbossammide (TT) in TT3: DOPE: Colesterolo: DMG-PEG2000 (1,2-dimiristoil-sn-glicerolo, metossipolietilenglicole) con un nucleo PLGA polimerico [160]. Inoltre, LPN ottimizzati costituiti dal polimero degradabile PBAE, formulato con PEG-Lipid C14-2000, hanno mostrato un rilascio riuscito dell’mRNA ai polmoni [161].
Questo, insieme alla co-consegna riportata di siRNA e mRNA con nanoparticelle ibride di polimeri lipidoidi [162], mostra che gli LPN sono un sistema emergente di rilascio di acido nucleico. La formulazione ibrida è termodinamicamente favorevole rispetto alle interazioni idrofobiche, di van der Waal ed elettrostatiche [80].
Diversi lipidi e polimeri sono stati studiati per formulare particelle lipidiche di acido nucleico stabili utilizzando questo sistema di rilascio. I polimeri più comuni impiegati sono PLGA, policaprolattone, acido polilattico o loro combinazioni, mentre i lipidi utilizzati includono DOTAP, 1,2-dilauroil-sn-glicero-3-fosfocolina, 1,2-distearoil-sn-glicero-3-fosfocolina , lecitina, DSPE e PEG, tra gli altri. Strutturalmente, sulla base della diffusione di raggi X a piccolo angolo e della microscopia elettronica a trasmissione criogenica, si suggerisce che queste nanoparticelle comportino un nucleo di matrice polimerica con strutture lipidiche lamellari con l’acido nucleico localizzato nel nucleo e nella corona [163].
Vettori a base di peptidi
I sistemi basati su peptidi per la somministrazione di mRNA stanno guadagnando slancio grazie alla versatilità che i peptidi possono offrire. In letteratura sono stati riportati sistemi di rilascio basati su peptidi, sia da soli che in combinazione con altri materiali come i polimeri. In uno studio riguardante la terapia del cancro ovarico, il peptide penetrante nelle cellule (CPP) disponibile in commercio PepFect14 è stato complessato con eGFP mRNA tramite interazioni elettrostatiche attraenti [164].
Questa formulazione nanoparticellare è stata più efficiente nella trasfezione dell’mRNA dell’eGFP in cellule associate al cancro ovarico rispetto alla lipofectamina MessengerMAX disponibile in commercio. Allo stesso modo, il CPP RALA è stato utilizzato per fornire efficacemente sia eGFP che OVA mRNA ed è stato dimostrato che supera il lipide cationico DOTAP e il lipide fusogenico DOPE [165]. Tuttavia, le attuali limitazioni includono il rilascio cellulare mirato [164] e una breve emivita di circolazione a causa della bassa stabilità nel mezzo contenente siero [166].
Recentemente, è stata introdotta una nuova nanoplatform di rilascio di mRNA ibrido polimero-peptide [167] combinando micelle a base di polimero (PLA) e un peptide fusogenico cationico (RALA) per ottenere degradabilità, stabilità dell’mRNA e proprietà endosomolitiche appropriate per la traduzione. È stato segnalato per proteggere eGFP e FLuc mRNA dalla degradazione della nucleasi sierica e ottenere la trasfezione DC. Infatti, i vettori e gli ibridi a base di peptidi sono aggiunte promettenti e interessanti ai vari vettori non virali esistenti per la consegna dell’mRNA.
Consegna dell’mRNA specifico delle cellule
La consegna cellulo-specifica dell’mRNA sarebbe utile per lo sviluppo di terapie basate sull’mRNA. Ciò può migliorare la consegna delle molecole di mRNA alle cellule bersaglio e quindi ridurre la dose di mRNA richiesta, oltre a ridurre i potenziali effetti fuori bersaglio. È stato riportato che gli organi linfoidi possono essere mirati regolando la carica netta della formulazione [177]. Ciò si basa sul principio che gli APC si trovano in prossimità dei linfociti T in questi organi, fornendo così condizioni ottimali per un priming efficiente e l’amplificazione delle risposte delle cellule T.
È stato dimostrato che il rilascio sito-specifico di nanoparticelle caricate con mRNA tramite targeting attivo provoca l’induzione di forti risposte effettrici e delle cellule T di memoria e la mediazione di un potente rigetto IFN-α-dipendente dei tumori progressivi, come osservato con gli RNA. In un altro studio, il rilascio cellulo-specifico di FLuc e IL-10 mRNA ai leucociti (Ly6c +) è stato ottenuto rivestendo gli LNP contenenti mRNA formulati con anticorpi monoclonali anti-L6c + [178].
In alternativa, DC e macrofagi esprimono recettori con la capacità di presentare antigeni, ad es., Recettori della lectina di tipo C [179], che riconoscono gruppi di zucchero come glicani terminati da mannosio e fucosio [180] e mediano l’endocitosi delle nanoparticelle modificate dal mannosio . Questo è stato sfruttato per la trasfezione dell’mRNA di GFP in DC mediante autoassemblaggio di coniugati mannosio-colesterolo con unità PEG variabili come linker [181].
link di riferimento: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7076378/
Ottimizzazione della traduzione e della stabilità dell’mRNA
Questo argomento è stato ampiamente discusso nelle revisioni precedenti14,15; quindi, riassumiamo brevemente i risultati chiave (Box 1). Gli elementi UTR 5 ′ e 3 ′ che fiancheggiano la sequenza codificante influenzano profondamente la stabilità e la traduzione dell’mRNA, entrambi aspetti critici per i vaccini.
Queste sequenze regolatorie possono essere derivate da geni virali o eucariotici e aumentare notevolmente l’emivita e l’espressione degli mRNA terapeutici23,24. Per una produzione efficiente di proteine dall’mRNA25 è necessaria una struttura a cappuccio da 5 ‘. Varie versioni di capsule da 5 ‘possono essere aggiunte durante o dopo la reazione di trascrizione utilizzando un enzima di copertura del virus del vaccinia26 o incorporando un cappuccio sintetico o analoghi del cappuccio anti-inversione27,28.
La coda poli (A) svolge anche un importante ruolo regolatore nella traduzione e stabilità dell’mRNA25; quindi, una lunghezza ottimale di poli (A) 24 deve essere aggiunta all’mRNA direttamente dal modello di DNA codificante o utilizzando la poli (A) polimerasi. L’utilizzo del codone ha inoltre un impatto sulla traduzione delle proteine.
La sostituzione di codoni rari con codoni sinonimi di uso frequente che hanno abbondante tRNA affine nel citosol è una pratica comune per aumentare la produzione di proteine dall’mRNA29, sebbene l’accuratezza di questo modello sia stata messa in dubbio30. L’arricchimento del contenuto di G: C costituisce un’altra forma di ottimizzazione della sequenza che ha dimostrato di aumentare i livelli di mRNA allo stato stazionario in vitro31 e l’espressione della proteina in vivo12.
Sebbene l’espressione della proteina possa essere modulata positivamente alterando la composizione del codone o introducendo nucleosidi modificati (discussi di seguito), è anche possibile che queste forme di ingegneria di sequenza possano influenzare la struttura secondaria dell’mRNA32, la cinetica e l’accuratezza della traduzione e del ripiegamento simultaneo delle proteine33,34 e l’espressione di epitopi criptici delle cellule T presenti in frame di lettura alternativi30. Tutti questi fattori potrebbero potenzialmente influenzare l’entità o la specificità della risposta immunitaria.
*-*-*-*-*-*-*-
Riquadro 1: Strategie per ottimizzare la farmacologia dell’mRNA
Attualmente vengono utilizzate numerose tecnologie per migliorare gli aspetti farmacologici dell’mRNA. Le varie modifiche dell’mRNA utilizzate e il loro impatto sono riassunte di seguito.
- • Gli analoghi del cappuccio sintetico e gli enzimi del capping26,27 stabilizzano l’mRNA e aumentano la traduzione delle proteine legandosi al fattore di inizio della traduzione eucariotica 4E (EIF4E)
- • Gli elementi regolatori nella regione 5′-untranslated (UTR) e 3′-UTR23 stabilizzano l’mRNA e aumentano la traduzione delle proteine
- • Poly (A) tail25 stabilizza l’mRNA e aumenta la traduzione delle proteine
- • I nucleosidi modificati9,48 riducono l’attivazione immunitaria innata e aumentano la traduzione
- • Tecniche di separazione e / o purificazione: il trattamento con RNasi III (NP e DW, osservazioni non pubblicate) e la purificazione mediante cromatografia liquida a proteine veloci (FPLC )13 riducono l’attivazione immunitaria e aumentano la traduzione
- • L’ottimizzazione della sequenza e / o del codone29 aumenta la traduzione
- • Modulazione delle cellule bersaglio: la co-consegna dei fattori di inizio della traduzione e altri metodi alterano la traduzione e l’immunogenicità
*-*-*-*-*-*-*-
Modulazione dell’immunogenicità
L’mRNA esogeno è intrinsecamente immunostimolante, poiché è riconosciuto da una varietà di recettori immunitari innati di superficie cellulare, endosomiali e citosolici (Fig. 1) (rivisti in Rif. 35). A seconda dell’applicazione terapeutica, questa caratteristica dell’mRNA potrebbe essere benefica o dannosa. È potenzialmente vantaggioso per la vaccinazione perché in alcuni casi può fornire un’attività adiuvante per guidare la maturazione delle cellule dendritiche (DC) e quindi suscitare robuste risposte immunitarie delle cellule T e B. Tuttavia, il rilevamento immunitario innato dell’mRNA è stato anche associato all’inibizione dell’espressione dell’antigene e può influenzare negativamente la risposta immunitaria9,13. Sebbene gli effetti paradossali del rilevamento immunitario innato su diversi formati di vaccini a mRNA siano compresi in modo incompleto, negli ultimi anni sono stati compiuti alcuni progressi nel chiarire questi fenomeni.
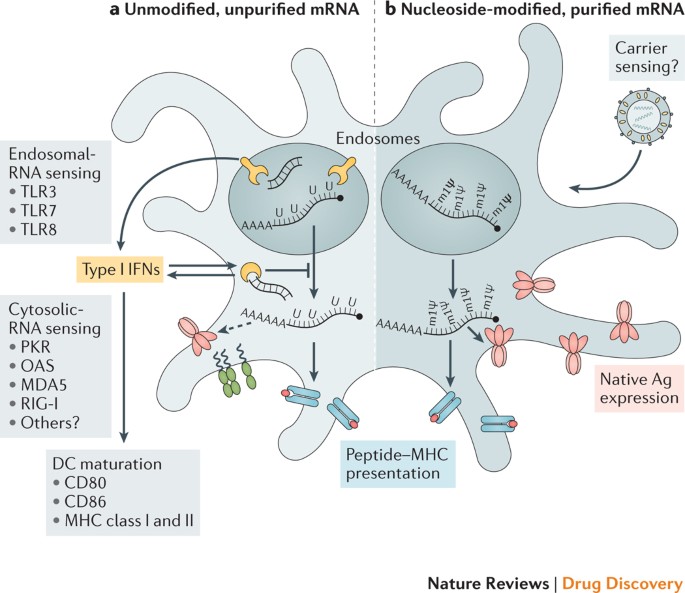
Studi dell’ultimo decennio hanno dimostrato che il profilo immunostimolante dell’mRNA può essere modellato dalla purificazione dell’mRNA IVT e dall’introduzione di nucleosidi modificati, nonché complessando l’mRNA con varie molecole trasportatrici9,13,36,37.
I preparati di mRNA sintetizzati enzimaticamente contengono contaminanti di RNA a doppio filamento (dsRNA) come prodotti aberranti della reazione IVT13. Come imitazione dei genomi virali e degli intermedi di replicazione, il dsRNA è un potente pattern molecolare associato a patogeni (PAMP) che viene rilevato dai recettori di riconoscimento del pattern in più compartimenti cellulari (Fig.1).
Il riconoscimento dell’mRNA IVT contaminato con dsRNA si traduce in una produzione robusta di interferone di tipo I13, che sovraregola l’espressione e l’attivazione della protein chinasi R (PKR; nota anche come EIF2AK2) e della 2′-5′-oligoadenilato sintetasi (OAS), determinando l’inibizione di traduzione38 e la degradazione dell’mRNA cellulare e dell’RNA ribosomiale39, rispettivamente.
Karikó e colleghi13 hanno dimostrato che il dsRNA contaminante può essere rimosso in modo efficiente dall’mRNA IVT mediante metodi cromatografici come la cromatografia liquida della proteina rapida in fase inversa (FPLC) o la cromatografia liquida ad alte prestazioni (HPLC). Sorprendentemente, la purificazione mediante FPLC ha dimostrato di aumentare la produzione di proteine dall’mRNA IVT fino a 1.000 volte nelle DC umane primarie13.
Pertanto, un’appropriata purificazione dell’mRNA IVT sembra essere fondamentale per massimizzare la produzione di proteine (immunogeni) nelle DC e per evitare l’attivazione immunitaria innata indesiderata.
Oltre ai contaminanti dsRNA, le molecole di mRNA a filamento singolo sono esse stesse un PAMP quando vengono fornite alle cellule in modo esogeno. Gli oligoribonucleotidi a filamento singolo ei loro prodotti degradativi vengono rilevati dai sensori endosomiali Toll-like receptor 7 (TLR7) e TLR8 (Refs 40,41), con conseguente produzione di interferone di tipo I42.
Fondamentalmente, è stato scoperto che l’incorporazione di nucleosidi modificati chimicamente presenti in natura, inclusi ma non limitati a pseudouridina9,43,44 e 1-metilpseudouridina45, impedisce l’attivazione di TLR7, TLR8 e altri sensori immunitari innati46,47, riducendo così il segnale dell’interferone di tipo I48 . La modifica dei nucleosidi sopprime anche parzialmente il riconoscimento delle specie di dsRNA46,47,48.
Di conseguenza, Karikó e altri hanno dimostrato che l’mRNA modificato con nucleosidi viene tradotto in modo più efficiente rispetto all’mRNA non modificato in vitro9, in particolare nelle DC primarie e in vivo nei topi45. In particolare, il livello più alto di produzione di proteine nelle DC è stato osservato quando l’mRNA era sia purificato con FPLC che modificato con nucleosidi13.
Questi progressi nella comprensione delle fonti del rilevamento immunitario innato e su come evitare i loro effetti avversi hanno contribuito in modo sostanziale all’attuale interesse per i vaccini a base di mRNA e le terapie sostitutive delle proteine.
In contrasto con i risultati sopra descritti, uno studio di Thess e colleghi ha scoperto che l’mRNA ottimizzato per sequenza, purificato da HPLC e non modificato produceva livelli più elevati di proteine nelle cellule HeLa e nei topi rispetto alla sua controparte modificata con nucleosidi12.
Inoltre, Kauffman e colleghi hanno dimostrato che l’mRNA non modificato e non purificato da HPLC ha prodotto una produzione di proteine più robusta nelle cellule HeLa rispetto all’mRNA modificato con nucleosidi e ha portato a livelli simili di produzione di proteine nei topi49.
Sebbene non del tutto chiare, le discrepanze tra i risultati di Karikó9,13 e questi autori12,49 potrebbero derivare da variazioni nell’ottimizzazione della sequenza di RNA, dalla severità della purificazione dell’mRNA per rimuovere i contaminanti del dsRNA e dal livello di rilevamento immunitario innato nei tipi cellulari mirati .
Le proprietà immunostimolatorie dell’mRNA possono invece essere aumentate mediante l’inclusione di un adiuvante per aumentare la potenza di alcuni formati di vaccino a mRNA. Questi includono adiuvanti tradizionali e nuovi approcci che sfruttano l’immunogenicità intrinseca dell’mRNA o la sua capacità di codificare le proteine immunomodulatorie.
I vaccini a RNA autoreplicante hanno mostrato una maggiore immunogenicità ed efficacia dopo la formulazione dell’RNA in una nanoemulsione cationica basata sull’adiuvante autorizzato MF59 (Novartis )50. Un’altra efficace strategia adiuvante è TriMix, una combinazione di mRNA che codificano tre proteine attivatrici immunitarie: CD70, CD40 ligando (CD40L) e TLR4 costitutivamente attivo.
L’mRNA TriMix ha aumentato l’immunogenicità dell’mRNA nudo, non modificato e non purificato in più studi sui vaccini contro il cancro ed è stato particolarmente associato con una maggiore maturazione delle DC e risposte citotossiche dei linfociti T (CTL) (rivisto in Ref.51). È stato inoltre dimostrato che il tipo di portatore di mRNA e la dimensione del complesso mRNA-vettore modulano il profilo delle citochine indotto dalla somministrazione di mRNA.
Ad esempio, la piattaforma vaccinale RNActive (CureVac AG )52,53 dipende dal suo vettore per fornire attività adiuvante. In questo caso, l’antigene è espresso da un mRNA nudo, non modificato, ottimizzato in sequenza, mentre l’attività adiuvante è fornita dall’RNA co-consegnato complessato con protamina (un peptide policationico), che agisce tramite il segnale TLR752,54.
Questo formato di vaccino ha suscitato risposte immunitarie favorevoli in molteplici studi preclinici su animali per la vaccinazione contro il cancro e le malattie infettive18,36,55,56. Un recente studio ha fornito informazioni meccanicistiche sull’adiuvanticità dei vaccini RNActive nei topi in vivo e nelle cellule umane in vitro54.
È stata dimostrata una potente attivazione di TLR7 (topo e umano) e TLR8 (umano) e produzione di interferone di tipo I, citochine proinfiammatorie e chemochine dopo l’immunizzazione intradermica54. Un’attività adiuvante simile è stata dimostrata anche nel contesto di vaccini non a base di mRNA utilizzando RNAdjuvant (CureVac AG), un RNA a filamento singolo non modificato stabilizzato da un peptide vettore cationico57.
Progressi nella consegna del vaccino mRNA
La consegna efficiente di mRNA in vivo è fondamentale per ottenere rilevanza terapeutica. L’mRNA esogeno deve penetrare la barriera della membrana lipidica per raggiungere il citoplasma per essere tradotto in proteina funzionale. I meccanismi di assorbimento dell’mRNA sembrano dipendere dal tipo di cellula e le proprietà fisico-chimiche dei complessi di mRNA possono influenzare profondamente il rilascio cellulare e la distribuzione degli organi.
Esistono due approcci di base per la consegna dei vaccini a mRNA che sono stati descritti fino ad oggi. In primo luogo, caricamento dell’mRNA nelle DC ex vivo, seguito dalla reinfusione delle cellule trasfettate58; e secondo, iniezione parenterale diretta di mRNA con o senza un vettore.
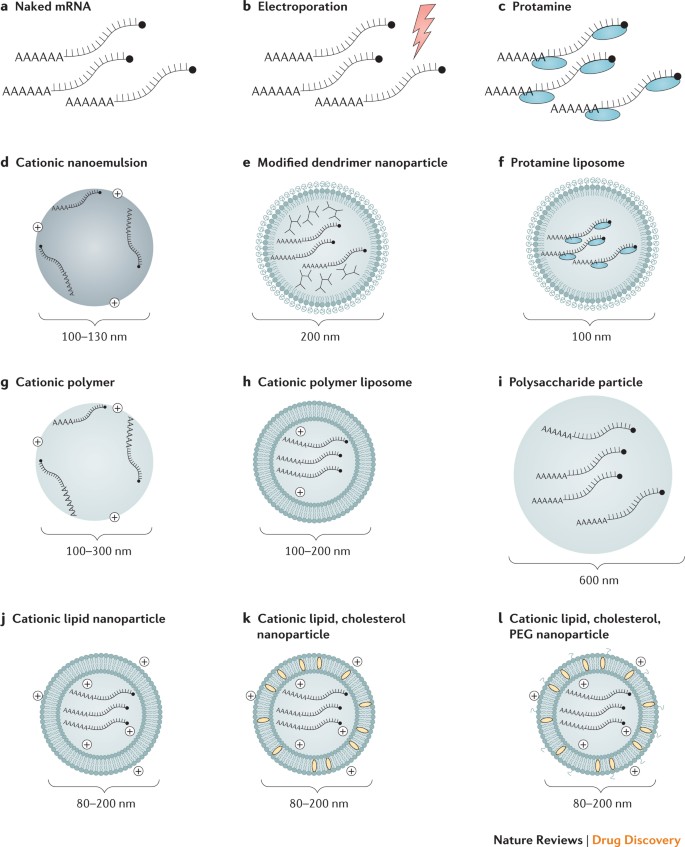
Il carico DC ex vivo consente un controllo preciso del bersaglio cellulare, dell’efficienza della trasfezione e di altre condizioni cellulari, ma come forma di terapia cellulare, è un approccio alla vaccinazione costoso e laborioso. L’iniezione diretta di mRNA è relativamente rapida ed economica, ma non consente ancora un rilascio specifico ed efficiente del tipo di cellula, sebbene ci siano stati progressi recenti in questo senso59. Entrambi questi approcci sono stati esplorati in una varietà di forme (Fig. 2; Tabella 1).
E x carico vivo delle DC . Le DC sono le cellule del sistema immunitario che presentano l’antigene più potenti. Iniziano la risposta immunitaria adattativa interiorizzando e processando proteoliticamente gli antigeni e presentandoli alle cellule T CD8 + e CD4 + sui principali complessi di istocompatibilità (MHC), vale a dire, MHC classe I e MHC classe II, rispettivamente. Inoltre, le DC possono presentare un antigene intatto ai linfociti B per provocare una risposta anticorpale60. Le DC sono anche altamente suscettibili alla trasfezione dell’mRNA. Per questi motivi, le DC rappresentano un bersaglio attraente per la trasfezione da vaccini a mRNA, sia in vivo che ex vivo.
Sebbene sia stato dimostrato che le DC interiorizzano l’mRNA nudo attraverso una varietà di vie endocitiche61,62,63, l’efficienza della trasfezione ex vivo è comunemente aumentata utilizzando l’elettroporazione; in questo caso, le molecole di mRNA passano attraverso i pori della membrana formati da un impulso ad alta tensione ed entrano direttamente nel citoplasma (rivisto in Ref. 64).
Questo approccio di consegna dell’mRNA è stato favorito per la sua capacità di generare un’elevata efficienza di trasfezione senza la necessità di una molecola vettore. Le DC caricate con mRNA ex vivo vengono quindi reinfuse nel destinatario del vaccino autologo per avviare la risposta immunitaria.
La maggior parte dei vaccini DC caricati ex vivo suscitano una risposta immunitaria prevalentemente cellulo-mediata; quindi, sono stati usati principalmente per trattare il cancro (rivisto in Ref. 58).
I njection di nuda mRNA in vivo. L’mRNA nudo è stato utilizzato con successo per le immunizzazioni in vivo, in particolare in formati che mirano preferenzialmente alle cellule presentanti l’antigene, come nelle iniezioni intradermiche61,65 e intranodali66,67,68.
In particolare, un recente rapporto ha mostrato che ripetute immunizzazioni intranodali con mRNA nudo e non modificato che codifica per neoantigeni associati al tumore hanno generato solide risposte delle cellule T e una maggiore sopravvivenza libera da progressione68 (discusso ulteriormente nel Box 2).
Metodi di consegna fisica in vivo.Per aumentare l’efficienza dell’assorbimento di mRNA in vivo, sono stati occasionalmente utilizzati metodi fisici per penetrare nella membrana cellulare. Un primo rapporto ha mostrato che l’mRNA complessato con particelle d’oro potrebbe essere espresso nei tessuti utilizzando una pistola genetica, un metodo microproiettile69.
La pistola genetica ha dimostrato di essere un metodo efficiente di somministrazione e vaccinazione dell’RNA nei modelli murini70,71,72,73, ma non sono disponibili dati di efficacia in animali di grandi dimensioni o nell’uomo. L’elettroporazione in vivo è stata utilizzata anche per aumentare l’assorbimento dell’RNA terapeutico74,75,76; tuttavia, in uno studio, l’elettroporazione ha aumentato l’immunogenicità solo di un RNA autoamplificante e non di un vaccino a base di mRNA non replicante74. I metodi fisici possono essere limitati da un aumento della morte cellulare e da un accesso limitato a cellule o tessuti bersaglio. Recentemente,
Protamine. È stato dimostrato che il peptide cationico protamina protegge l’mRNA dalla degradazione da parte delle RNasi sieriche77; tuttavia, il solo mRNA complessato con protamina ha dimostrato un’espressione proteica ed efficacia limitate in un modello di vaccino contro il cancro, probabilmente a causa di un’associazione eccessivamente stretta tra protamina e mRNA36,78. Questo problema è stato risolto sviluppando la piattaforma vaccinale RNActive, in cui l’RNA formulato con protamina serve solo come attivatore immunitario e non come vettore di espressione52.
Lipidi cationici e rilascio a base di polimeri.Reagenti di trasfezione di mRNA altamente efficienti a base di lipidi o polimeri cationici, come TransIT-mRNA (Mirus Bio LLC) o Lipofectamine (Invitrogen), sono disponibili in commercio e funzionano bene in molte cellule primarie e linee cellulari tumorali9,13, ma spesso mostrano un numero limitato di efficacia in vivo o un alto livello di tossicità (NP e DW, osservazioni non pubblicate).
Sono stati compiuti grandi progressi nello sviluppo di reagenti complessanti progettati in modo simile per un uso sicuro ed efficace in vivo, e questi sono discussi in dettaglio in diverse revisioni recenti10,11,79,80. Lipidi cationici e polimeri, compresi i dendrimeri, sono diventati strumenti ampiamente utilizzati per la somministrazione di mRNA negli ultimi anni.
Il campo dell’mRNA ha chiaramente beneficiato del sostanziale investimento nella somministrazione in vivo di piccoli RNA interferenti (siRNA), dove questi veicoli per le consegne sono stati utilizzati per oltre un decennio. Le nanoparticelle lipidiche (LNP) sono diventate uno degli strumenti di rilascio di mRNA più attraenti e comunemente usati.
Gli LNP sono spesso costituiti da quattro componenti: un lipide cationico ionizzabile, che promuove l’autoassemblaggio in particelle delle dimensioni di un virus (~ 100 nm) e consente il rilascio endosomiale di mRNA al citoplasma; polietilenglicole lipidico (PEG), che aumenta l’emivita delle formulazioni; colesterolo, un agente stabilizzante; e fosfolipidi presenti in natura, che supportano la struttura del doppio strato lipidico.
Numerosi studi hanno dimostrato un’efficace consegna di siRNA in vivo da parte degli LNP (rivisto in Ref.81), ma solo di recente è stato dimostrato che gli LNP sono potenti strumenti per il rilascio in vivo di RNA19 autoamplificante e mRNA21 convenzionale non replicante.
È stato dimostrato che i complessi mRNA-LNP somministrati sistemicamente prendono di mira principalmente il fegato a causa del legame dell’apolipoproteina E e del successivo assorbimento mediato dal recettore da parte degli epatociti82, e la somministrazione intradermica, intramuscolare e sottocutanea ha dimostrato di produrre un’espressione proteica prolungata nel sito di iniezione21,22.
I meccanismi di fuga dell’mRNA nel citoplasma non sono completamente compresi, non solo per i liposomi artificiali ma anche per gli esosomi presenti in natura83. Ulteriori ricerche in quest’area saranno probabilmente di grande beneficio nel campo della somministrazione terapeutica dell’RNA. I meccanismi di fuga dell’mRNA nel citoplasma non sono completamente compresi, non solo per i liposomi artificiali ma anche per gli esosomi naturali83.
Ulteriori ricerche in quest’area saranno probabilmente di grande beneficio nel campo della somministrazione terapeutica dell’RNA. I meccanismi di fuga dell’mRNA nel citoplasma non sono completamente compresi, non solo per i liposomi artificiali ma anche per gli esosomi naturali83. Ulteriori ricerche in quest’area saranno probabilmente di grande beneficio nel campo della somministrazione terapeutica dell’RNA.
L’entità e la durata della produzione di proteine in vivo dai vaccini mRNA-LNP possono essere controllate in parte variando la via di somministrazione. È stato dimostrato che il rilascio intramuscolare e intradermico di mRNA-LNP produce un’espressione proteica più persistente rispetto alle vie di consegna sistemiche: in un esperimento, l’emivita della luciferasi di lucciola codificata da mRNA era circa tre volte più lunga dopo l’iniezione intradermica rispetto a quella dopo la consegna endovenosa21.
Queste cinetiche dell’espressione di mRNA-LNP possono essere favorevoli per indurre risposte immunitarie. Uno studio recente ha dimostrato che la disponibilità sostenuta dell’antigene durante la vaccinazione era un fattore trainante di titoli anticorpali elevati e risposte delle cellule B del centro germinale (GC) e delle cellule T helper follicolare (TFH )84.
Questo processo è stato potenzialmente un fattore che contribuisce alla potenza dei vaccini mRNA-LNP modificati con nucleosidi recentemente descritti, somministrati per via intramuscolare e intradermica20,22,85. In effetti, le cellule TFH sono state identificate come una popolazione critica di cellule immunitarie che i vaccini devono attivare per generare risposte anticorpali neutralizzanti potenti e di lunga durata, in particolare contro i virus che eludono l’immunità umorale86.
Le dinamiche della reazione GC e la differenziazione delle cellule TFH sono comprese in modo incompleto e i progressi in queste aree sarebbero senza dubbio fruttuosi per la progettazione di vaccini futuri (Box 3). Le cellule TFH sono state identificate come una popolazione critica di cellule immunitarie che i vaccini devono attivare per generare risposte anticorpali neutralizzanti potenti e di lunga durata, in particolare contro i virus che eludono l’immunità umorale86.
Le dinamiche della reazione GC e la differenziazione delle cellule TFH sono comprese in modo incompleto e i progressi in queste aree sarebbero senza dubbio fruttuosi per la progettazione di vaccini futuri (Box 3). Le cellule TFH sono state identificate come una popolazione critica di cellule immunitarie che i vaccini devono attivare per generare risposte anticorpali neutralizzanti potenti e di lunga durata, in particolare contro i virus che eludono l’immunità umorale86. Le dinamiche della reazione GC e la differenziazione delle cellule TFH sono comprese in modo incompleto e i progressi in queste aree sarebbero senza dubbio fruttuosi per la progettazione di vaccini futuri (Box 3).
*-*-*-*-*-
Riquadro 2: vaccini contro il cancro neoepitopi personalizzati
Sahin e colleghi hanno sperimentato l’uso di vaccini contro il cancro individualizzati del neoepitopo mRNA121. Usano il sequenziamento ad alto rendimento per identificare ogni mutazione somatica unica del campione di tumore di un singolo paziente, chiamato mutanoma.
Ciò consente la progettazione razionale dei vaccini contro il cancro neoepitopo in modo specifico per il paziente e ha il vantaggio di mirare a specificità antigeniche non auto che non dovrebbero essere eliminate dai meccanismi di tolleranza centrale. Recentemente è stata fornita una dimostrazione del concetto: Kreiter e colleghi hanno scoperto che una parte sostanziale delle mutazioni tumorali non sinonime erano immunogeniche quando rilasciate dall’mRNA ed erano principalmente riconosciute dalle cellule T CD4 +176.
Sulla base di questi dati, hanno generato un metodo computazionale per prevedere i neoepitopi di classe II limitati al complesso maggiore di istocompatibilità (MHC) che possono essere utilizzati come immunogeni del vaccino. I vaccini a mRNA che codificano tali neoepitopi hanno controllato la crescita del tumore nel melanoma B16-F10 e nei modelli murini di cancro del colon CT26.
In un recente studio clinico, Sahin e colleghi hanno sviluppato vaccini mRNA personalizzati basati su neoepitopi per 13 pazienti con melanoma metastatico, un cancro noto per la sua alta frequenza di mutazioni somatiche e quindi neoepitopi. Hanno immunizzato contro dieci neoepitopi per individuo iniettando mRNA nudo per via intranodale.
Le risposte delle cellule T CD4 + sono state rilevate contro la maggior parte dei neoepitopi ed è stata osservata una bassa frequenza di malattia metastatica dopo diversi mesi di follow-up68. È interessante notare che risultati simili sono stati ottenuti anche in uno studio di disegno analogo che utilizzava peptidi sintetici come immunogeni piuttosto che mRNA177. Insieme, questi recenti studi suggeriscono la potenziale utilità della metodologia del vaccino personalizzato.
*-*-*-*-*-
Riquadro 3: il centro germinale e le cellule T helper follicolari
La stragrande maggioranza dei potenti vaccini antimicrobici induce risposte anticorpali protettive di lunga durata contro il patogeno bersaglio. Gli anticorpi ad alta affinità vengono prodotti in siti microanatomici specializzati all’interno dei follicoli delle cellule B degli organi linfoidi secondari chiamati centri germinali (GC).
La proliferazione delle cellule B, l’ipermutazione somatica e la selezione di mutanti ad alta affinità si verificano nei GC e per questi processi è necessario un aiuto efficiente delle cellule T178. La caratterizzazione della relazione tra GC B e cellule T è stata attivamente studiata negli ultimi anni. Il recettore follicolare homing recettore CXC-chemochine 5 (CXCR5) è stato identificato sulle cellule GC B e T negli anni ‘90179,180, ma il concetto di una linea specifica di cellule T helper follicolari (TFH) non è stato proposto fino al 2000 (Rif 181, 182).
L’esistenza del lignaggio TFH è stata confermata nel 2009 quando è stato identificato il fattore di trascrizione specifico per le cellule TFH, la proteina del linfoma 6 delle cellule B (BCL-6 )183,184,185. Le cellule TFH rappresentano un sottoinsieme specializzato di cellule T CD4 + che producono segnali critici per la sopravvivenza, la proliferazione e la differenziazione delle cellule B oltre a segnali per la commutazione isotipica degli anticorpi e per l’introduzione di mutazioni diversificanti nei geni delle immunoglobuline.
Le principali citochine prodotte dalle cellule TFH sono l’interleuchina-4 (IL-4) e IL-21, che svolgono un ruolo chiave nel guidare la reazione GC. Altri importanti marcatori e ligandi funzionali espressi dalle cellule TFH includono il ligando CD40 (CD40L), la proteina 1A (SH2D1A) contenente il dominio di omologia Src 2 (SH2), la proteina di morte cellulare programmata 1 (PD1) e il co-stimolatore delle cellule T inducibili (ICOS ) 186. La caratterizzazione di anticorpi anti-HIV-1 rari e ampiamente neutralizzanti ha rivelato che tassi insolitamente elevati di ipermutazione somatica sono un segno distintivo delle risposte anticorpali protettive contro l’HIV-1 (Rif. 187).
Poiché le cellule TFH svolgono un ruolo chiave nel guidare questo processo nelle reazioni GC, è urgentemente necessario lo sviluppo di nuovi adiuvanti o piattaforme vaccinali in grado di attivare in modo potente questo tipo di cellule.
link di riferimento: https://www.nature.com/articles/nrd.2017.243



{kind=link}