









 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Scientists have known for a while that SARS-CoV-2’s distinctive “spike” proteins help the virus infect its host by latching on to healthy cells. Now, a major new study shows that they also play a key role in the disease itself.
The paper, published on April 30, 2021, in Circulation Research, also shows conclusively that COVID-19 is a vascular disease, demonstrating exactly how the SARS-CoV-2 virus damages and attacks the vascular system on a cellular level.
The findings help explain COVID-19’s wide variety of seemingly unconnected complications, and could open the door for new research into more effective therapies.
“A lot of people think of it as a respiratory di
sease, but it’s really a vascular disease,” says Assistant Research Professor Uri Manor, who is co-senior author of the study. “That could explain why some people have strokes, and why some people have issues in other parts of the body. The commonality between them is that they all have vascular underpinnings.”
Salk researchers collaborated with scientists at the University of California San Diego on the paper, including co-first author Jiao Zhang and co-senior author John Shyy, among others.
While the findings themselves aren’t entirely a surprise, the paper provides clear confirmation and a detailed explanation of the mechanism through which the protein damages vascular cells for the first time.
There’s been a growing consensus that SARS-CoV-2 affects the vascular system, but exactly how it did so was not understood. Similarly, scientists studying other coronaviruses have long suspected that the spike protein contributed to damaging vascular endothelial cells, but this is the first time the process has been documented.
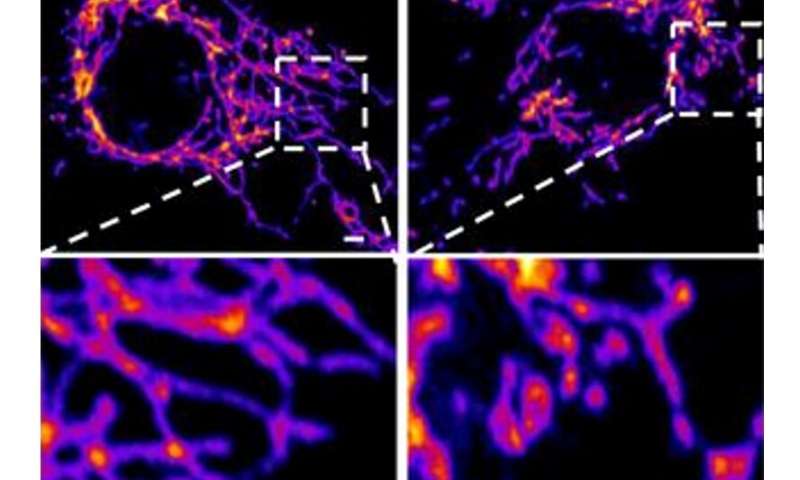
In the new study, the researchers created a ‘pseudovirus’ that was surrounded by SARS-CoV-2 classic crown of spike proteins, but did not contain any actual virus. Exposure to this pseudovirus resulted in damage to the lungs and arteries of an animal modelproving that the spike protein alone was enough to cause disease. Tissue samples showed inflammation in endothelial cells lining the pulmonary artery walls.
The team then replicated this process in the lab, exposing healthy endothelial cells (which line arteries) to the spike protein. They showed that the spike protein damaged the cells by binding ACE2.
This binding disrupted ACE2’s molecular signaling to mitochondria (organelles that generate energy for cells), causing the mitochondria to become damaged and fragmented.
Previous studies have shown a similar effect when cells were exposed to the SARS-CoV-2 virus, but this is the first study to show that the damage occurs when cells are exposed to the spike protein on its own.
“If you remove the replicating capabilities of the virus, it still has a major damaging effect on the vascular cells, simply by virtue of its ability to bind to this ACE2 receptor, the S protein receptor, now famous thanks to COVID,” Manor explains. “Further studies with mutant spike proteins will also provide new insight towards the infectivity and severity of mutant SARS CoV-2 viruses.”
As humans age, the speed and strength of their immune response weaken due to the loss of certain immune tissues such as the thymus, as well as poorer energy metabolism at the cellular level with mitochondria. The energy in the cell comes in the form of adenosine triphosphate (ATP) and is made by mitochondria when the cell is fueled with oxygen, as well as by glycolysis in the absence of oxygen.
Mitochondria are also known to interact with viral particles when they infect human host cells, engaging interferon and cytokine release, stimulating inflammation, and influencing viral survival and replication (Khan et al., 2015; Tiku et al., 2020). Studying mitochondrial-based immunity against the SARS-CoV-2 may give insight into why older individuals, with lessened mitochondrial efficiency, maybe worse equipped to face COVID-19.
The purpose of our study is to explore the molecular link between mitochondria in aged individuals and SARS-CoV-2. Furthermore, we also highlighted the role of various age-related comorbidities such as diabetes, obesity, and neurological illnesses in increased mortality rates amongst the elderly with COVID-19. We also explore current treatments, lifestyles, and safety measures that can help protect against COVID-19.
Mitochondria and Immunity
Mitochondria are organelles with a double membrane that serves as a cell’s primary source of energy production in the form of ATP and contribute to homeostasis, cell proliferation, cell death, and synthesis of amino acids, lipids, and nucleotides. In the event of an infection, mitochondria contribute to immunity by engaging the interferon system, altering their structure, and inducing programmed cell death (apoptosis; Ohta and Nishiyama, 2011).
Interferon Signaling and Mitochondria
Upon viral infection, the host’s innate immune system recognizes certain patterns, such as viral nucleic acid sequences or viral proteins, when they attach to receptors on host cellular membranes, intracellularly and extracellularly. Their recognition activates signaling pathways that lead to the inflammatory response. There are different types of receptors that these viral components attach to, including toll-like receptors and retinoic acid-inducible gene-I-like receptors (RLRs; Takeuchi and Akira, 2009).
Toll-like receptors are involved in activating type-I interferon, an inflammatory cytokine, and chemokine production and are found on the cell surface, endosome, and endoplasmic reticulum membranes (Takeuchi and Akira, 2009). RLRs are the cytosolic receptors that start the production of type-I interferon in nonimmune cells (Takeuchi and Akira, 2009) and can be found on mitochondria (Tal and Iwasaki, 2009).
These RLRs detect viral RNA in the cytoplasm and are involved in the recognition of RNA viruses such as paramyxoviruses, Japanese encephalitis virus, influenza virus, and picornaviruses (Kato et al., 2006). However, certain subtypes of RLRs may be involved in the detection of certain DNA viruses as well. For example, adenovirus and Herpes Simplex Virus Type 1 have a DNA-dependent RNA polymerase III that affects RLRs and stimulates interferon-β production, and the Epstein-Barr virus produces small RNA fragments that can activate RLRs (Samanta et al., 2006; Cheng et al., 2007; Chiu et al., 2009).
A component of the signaling pathway stemming from RLR activation is a molecule known as mitochondrial antiviral signaling protein (MAVS; Kawai et al., 2005). MAVS is located on the outer mitochondrial membrane (OMM; Seth et al., 2005) and upon activation, triggers transcription factors that will result in additional interferon production (Zhang et al., 2014). In addition to MAVS, the mitochondria-associated protein called “stimulator of interferon genes” and the mitochondrial protein mitofusin 2 are also involved in RLR cascades or work with MAVS (Ishikawa and Barber, 2008; Yasukawa et al., 2009). This evidence goes to show that mitochondria are an important part of interferon signaling in the immune system. Some viruses alter MAVS levels to prevent interferon production; the influenza A virus (Varga et al., 2012), the measles virus (Xia et al., 2014), the Newcastle disease virus (Meng et al., 2014), and the Hepatitis C virus (Ohta and Nishiyama, 2011) reduce or degrade MAVS as a way to prolong survival by reducing interferon signaling.
Mitochondrial Fission and Fusion Modulation
Viruses can manipulate mitochondrial fission and fusion to benefit viral survival (Holder and Reddy, 2020). Mitochondria can alter their structure through fission and fusion of their OMM and inner mitochondrial membrane (IMM), by functions involving GTPases related to dynamin (Tiku et al., 2020). The fusion of the OMM is mediated by proteins Mitofusin 1 and Mitofusin 2 via GTP hydrolysis, and fusion of the IMM is mediated by the protein optic atrophy 1, which is a GTPase present in the IMM (Tiku et al., 2020). Mitochondrial fusion is needed for the exchange of mitochondrial DNA, proteins, and metabolites (Archer, 2013).
On the other hand, fission of the OMM is mediated by the cytosolic GTPase dynamin-related protein 1 (Drp1) via GTP hydrolysis (Mears et al., 2011). Upon finding a mitochondrial scission site, Drp1 interacts with mitochondrial fission factor and mitochondrial dynamics proteins 49 and 51 to constrict and cut the OMM (Mears et al., 2011; Loson et al., 2013). The mechanisms of mitochondrial fission are not well understood. Fission is needed for removing damaged parts of mitochondria to be cleared by mitophagy (autophagy of the mitochondria) and is needed during cell cycle replication (Mao and Klionsky, 2013). Thus, enhanced fission usually leads to increased mitophagy.
Some viruses may promote mitochondrial fusion to reduce the interferon pro-inflammatory response against viruses through a mechanism that involves mitofusin 2 inhibition of MAVS. For example, the dengue virus stimulates mitochondrial fusion via its nonstructural protein NS4B (Barbier et al., 2017), and HIV enhances fusion via its envelope protein gp120 (Fields et al., 2016).
The SARS coronavirus (SARS-CoV-1) enhances fusion via its virulence factor ORF-9b (Shi et al., 2014). These virulence factors reduce the levels of Drp1, the fission-inducing protein, thus leading to unbalanced mitochondrial fusion, which is driven by mitofusin 2. As mitofusin 2 interacts with and inhibits MAVS, which typically increases interferons (Yasukawa et al., 2009) this can hinder the interferon response. Interestingly, SARS-CoV-1 uses the same ORF-9b to also reduce levels of MAVS directly, which further lowers the interferon response (Shi et al., 2014).
Some viruses induce mitochondrial fission to enhance mitophagy and alter the rate of apoptosis, usually via up-regulation or activation of Drp1 and/or degradation or inhibition of MAVS (Khan et al., 2015). Among these is the Hepatitis C virus via its core proteins and proteins E1-E2 (Kim et al., 2014), the Human cytomegalovirus via viral protein vMIA (McCormick et al., 2003), and the Hepatitis B virus via viral protein HBx (Kim et al., 2013). Fission mediated by these three particular viruses leads to inhibition of apoptosis so that the virus may survive for longer and further replicate (McCormick et al., 2003; Kim et al., 2013, 2014).
Due to the modulation by viruses on mitochondrial fusion and fission, their presence may lead to altered energy levels by way of mitochondrial count and form. Viruses that cause mitochondrial fission and lead to inhibition of apoptosis can allow viral particles to survive unharmed for longer. Many patients often feel weak from a lack of energy when infected with a viral illness. This may be due to the poorer mitochondrial energy production as a result of the increased fission.
Cell Death
Apoptosis, or programmed cell death, is another important function of the cell influenced by the mitochondria. There is an extrinsic pathway to activate apoptosis, controlled by certain ligands binding to “death” receptors, and an intrinsic pathway that is controlled by mitochondria (Brenner and Mak, 2009).
In this intrinsic pathway, the mitochondrial membrane is permeated and the mitochondrial membrane potential (MMP) is disrupted as the intermembrane space proteins spill into the cytoplasm (Shawgo et al., 2008). These proteins include cytochrome c (Liu et al., 1996), caspase-9 (Du et al., 2000), and apoptosis protease activating factor 1 that work together to form an apoptosome, which stimulates the final caspases to carry out cell death procedure (Cain et al., 2002).
MMP destabilization, which leads to apoptosis, is thought to occur by various mechanisms. First, there is the mechanism of selective OMM permeabilization (Kuwana et al., 2002). Bax and Bak, proteins that make up the Bax proapoptotic subfamily of Bcl-2 proteins, serve as pores on the mitochondrial membrane to maintain the MMP as well as release cytochrome c and calcium from within the mitochondria (Nutt et al., 2002).
When another proapoptotic subfamily known as the BH3-only subfamily attaches to activated Bax/Bak, they enhance permeability and increase the chance of apoptosis (Chen et al., 2005). On the other hand, the antiapoptotic Bcl-2 subfamily (including Bcl-2 and Bcl-xL) can attach to activated Bax/Bak proteins and inhibit Bax/Bak by forming an antiapoptotic complex and leading to decreased apoptosis (Shawgo et al., 2008).
A second mechanism involves the lipid bilayer makeup of the mitochondrial membrane and involves Bax creating rapid reorganization of the lipids that leads to structural stress and hole formation (Terrones et al., 2004). The last mechanism involves the stimulation of the permeability transition pore complex (PTPC) in the IMM (Shimizu et al., 1999). This is triggered by an overabundance of calcium or reactive oxygen species (Deniaud et al., 2008) and can be influenced by proteins from all parts of the mitochondria.
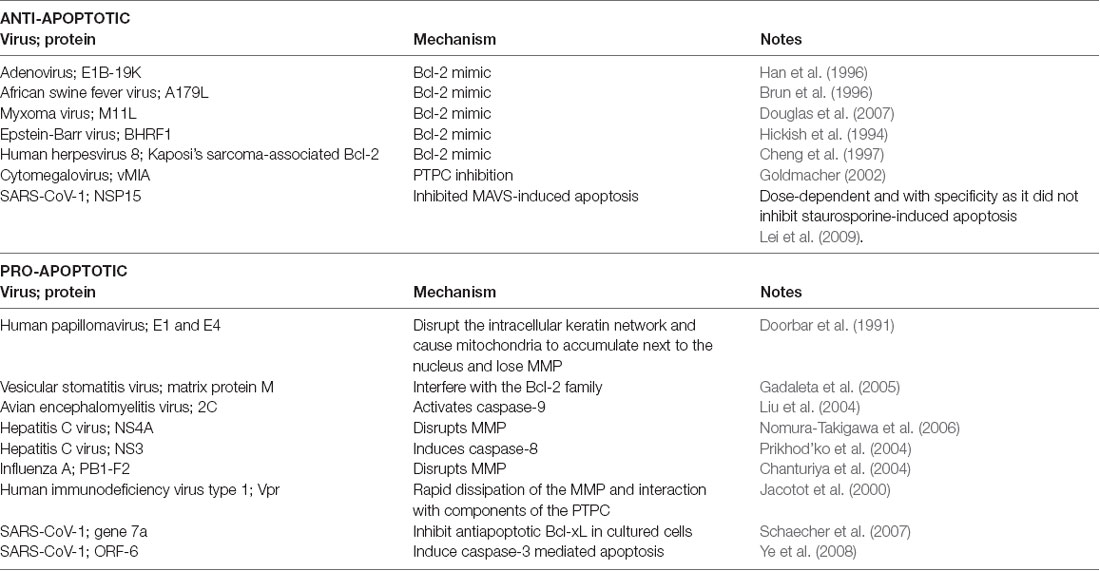
Viruses can use viral proteins that mimic Bcl-2 family members and other factors that are involved in the apoptosis pathways to manipulate the cell’s lifespan as they see fit. Table 1 demonstrates some common examples to illustrate this point.
SARS-CoV-2 and Mitochondria
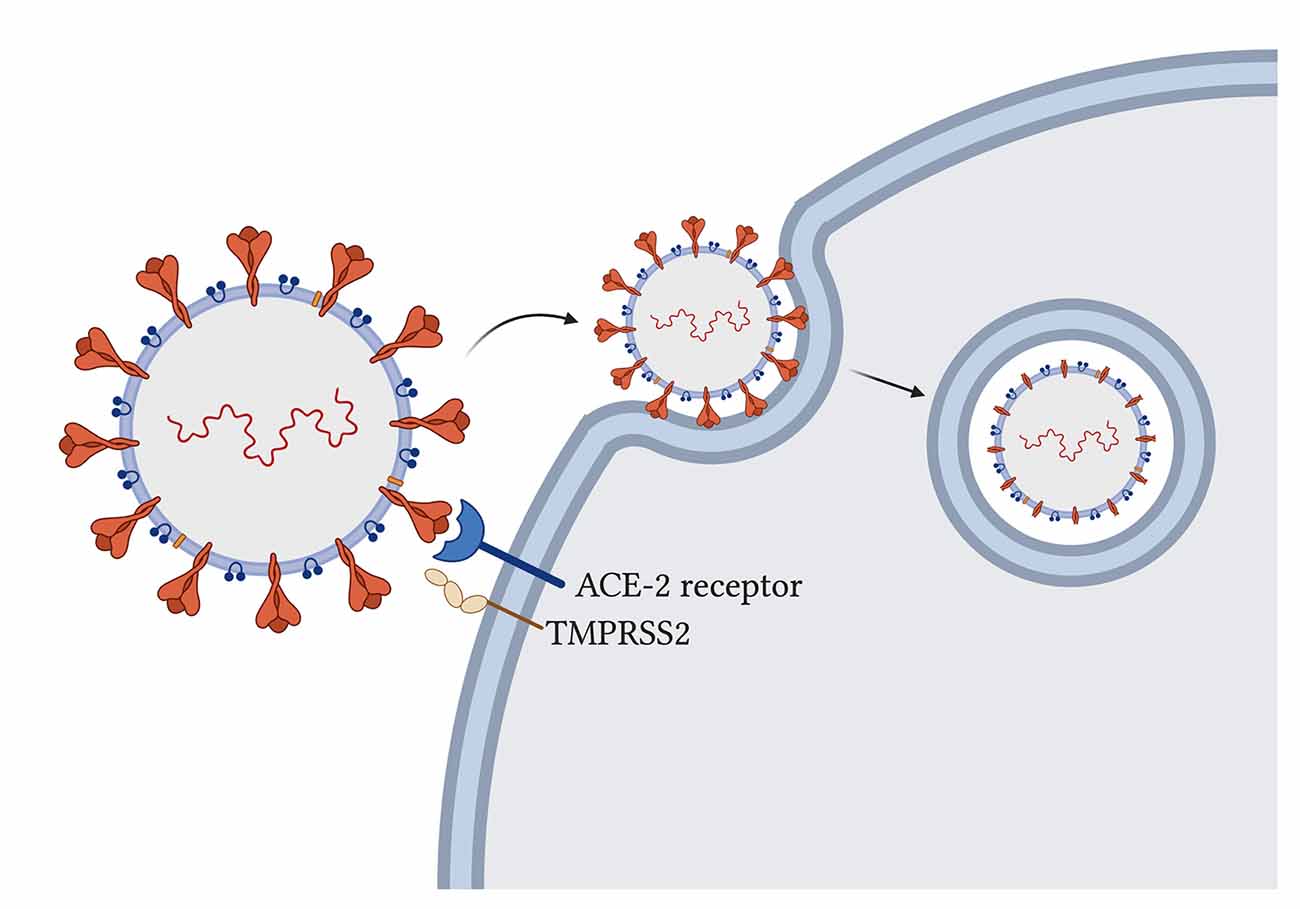
The novel SARS-CoV-2 uses its spike glycoprotein on the angiotensin-converting enzyme-2 (ACE-2) host receptor (Cao et al., 2020) to enter human host cells and host transmembrane serine protease 2 (TMPRSS2) to prime the spike protein for attachment (Hoffmann et al., 2020; Figure 2).
The virus particle enters the cell via endocytosis, and it has been proposed that the spike protein needs to be cleaved by host enzymes for viral entry to take place (Ou et al., 2020). ACE-2 influences mitochondrial functions and a lack of ACE-2 correlates with decreased ATP production and altered activation of NADPH oxidase 4 in the mitochondria, which is normally used for ROS production (Singh et al., 2020) that can both protect the cell by destroying pathogens or trigger the infected cell to go into apoptosis.
With the SARS-CoV-2 virus using ACE-2 receptors for its entry, the availability of ACE-2 for its usual functions may be impaired and contribute to symptom development.
Additionally, some studies have suggested that the TMPRSS2 from SARS-CoV-2 also influences mitochondrial function by acting on the estrogen-related receptor alpha, which is a nuclear receptor that regulates transcription of mitochondrial functions and energy homeostasis (Xu et al., 2018; Hoffmann et al., 2020; Singh et al., 2020).
Once inside the cell, SARS-CoV-2 triggers a massive inflammatory response. Through the innate immunity functions triggered upon viral infection detailed above, cytokines such as TNF-α, INF-γ, and interleukin-10 arrive at infected cells and cause an increase in mitochondrial ROS production through gene expression upregulation and electron transport chain modulation (Saleh et al., 2020).
Mitochondrial ROS then stimulates additional proinflammatory cytokine production (Li et al., 2013) as the virus continues to persist, eventually leading to a “cytokine storm” in which over-inflammation can cause fatal harm if adaptive immunity does not take over in time.
The immune response also causes the mitochondria to divert some energy away from ATP production to contribute to ROS production, which can harm the mitochondria in overwhelming amounts, leading to membrane permeabilization and apoptosis (Saleh et al., 2020). If severely damaged mitochondria release their contents into the cytosolic space, they stimulate the production of more cytokines such as IL-1β and IL-6 which are hallmarks for COVID-19 (Saleh et al., 2020).
Another mechanism of mitochondrial disruption employed by SARS-CoV-2 involves ferritin as evidenced by the high levels of ferritin in those with severe outcomes (Aguirre and Culotta, 2012). A normally functioning mitochondrion uses this iron to make heme, create iron-sulfur clusters, and store as mitochondrial ferritin (Saleh et al., 2020), but an overload of iron can lead to oxidative stress and impair mitochondrial function by reducing oxygen consumption by the mitochondria (Tang et al., 2020). Additionally, the ferritin overload can disrupt glucose tolerance in these cells with mitochondrial oxidative stress (Tang et al., 2020), which has implications for diabetic patients.
It is theorized that SARS-CoV-2 uses double-membrane vesicles derived from mitochondrial membranes to hide and protect itself inside the cell (Singh et al., 2020). This theory is based on evidence of HIV using ER-derived double-membrane vesicles (Somasundaran et al., 1994) and an observation that a point mutation in the coronavirus in rodents was shown to decrease ER-derived vesicles and increase localization of the virus to mitochondria at the same time (Clementz et al., 2008).
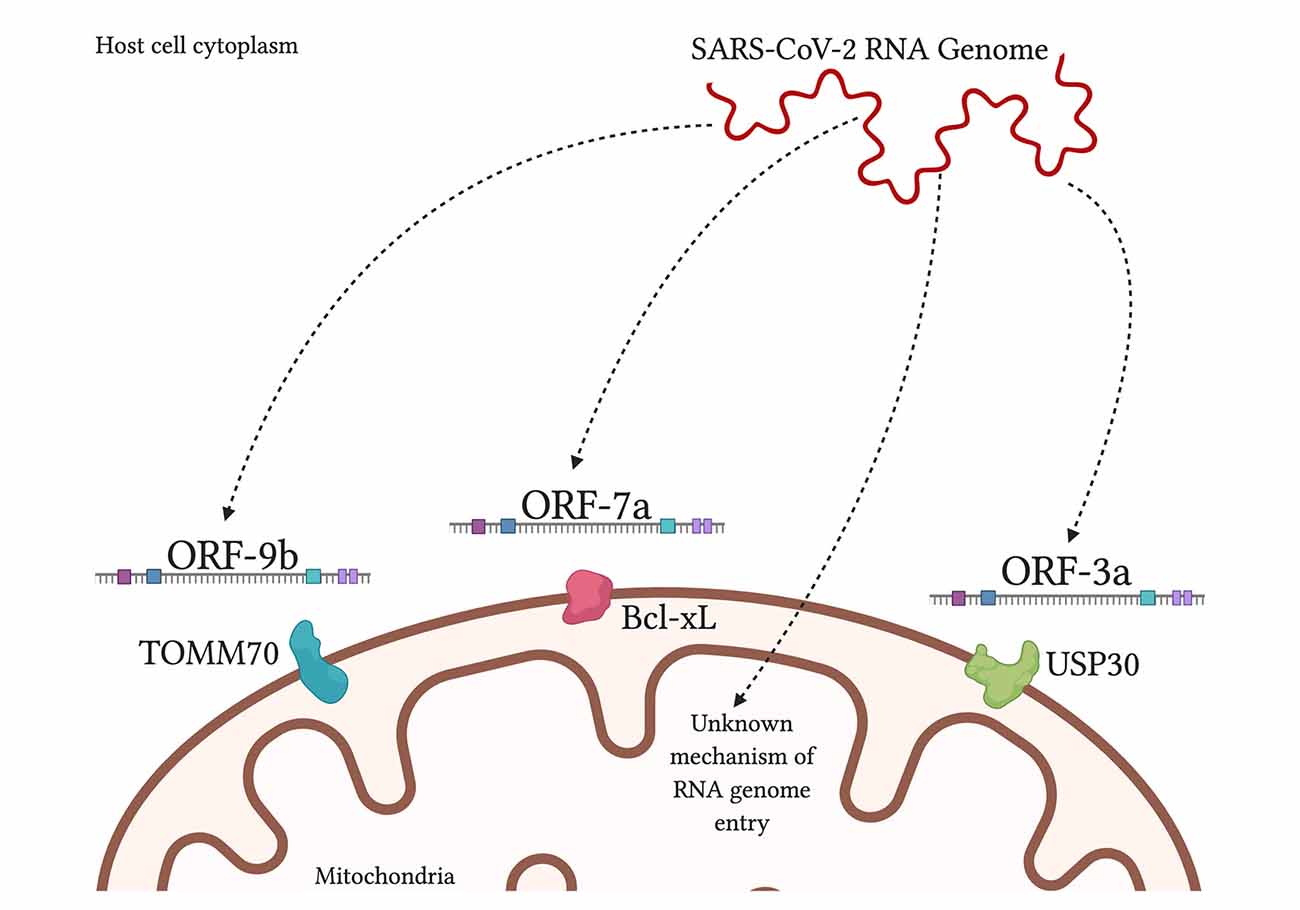
Furthermore, a study found 5′ and 3′ untranslated regions on SARS-CoV-2 unique for mitochondrial localization, although further work needs to be done on this finding (Wu et al., 2020). When comparing SARS-CoV-1 and SARS-CoV-2, both are found to contain open reading frame ORF-9b, ORF-7a, and ORF-8b, which localize to the mitochondria, in the case of SARS-CoV-1, to alter MAVS function and mitochondrial function (Chen et al., 2007; Shi et al., 2014).
SARS-CoV-2 additionally had ORF-3a present but ORF-3b absent (Singh et al., 2020; Figure 3). By encouraging the formation of double-membrane vesicles from the mitochondrial membrane or even the ER membrane, SARS-CoV-2 can safely avoid attacks from ROS and host proteases that threaten its survival. Meanwhile, the ROS is lingering around and can attack healthy tissue.
There is evidence of the SARS-CoV-1 ORF-9b causing mitochondrial fusion by the degradation of Drp1 by proteasomes (Shi et al., 2014; Holder and Reddy, 2020), and given the similarities in the genome, it is likely that the SARS-CoV-2 ORF-9b is lowering the amount of Drp1 as well, leading to more fusion. Mitochondrial fusion, which partly occurs via mitofusin 2, may lead to a hindered interferon response via inhibition of MAVS (Yasukawa et al., 2009).
While this suggests that the lowered interferon numbers may take away from interferon-induced apoptosis specifically (Chawla-Sarkar et al., 2003), we must consider that SARS-CoV-1 is known to induce apoptosis via other factors such as ORF-6 and -7a (Schaecher et al., 2007; Ye et al., 2008). Comparing both SARS viruses indicates that SARS-CoV-2 may induce apoptosis when its need for the human host cell is over. Additionally, there is some evidence for ferroptosis or ferritin-induced apoptosis with iron overload. Defective mitochondria cannot metabolize iron as they normally would, leading to iron buildup and ferroptosis (Saleh et al., 2020). This all implies a greater number of cell death with COVID-19.
SARS-CoV-2 may also interfere with platelet count and coagulation, specifically with increasing coagulability and decreasing platelet count as the severity of COVID-19 increases (Tang et al., 2020; Terpos et al., 2020). Apart from the increasing risk of stroke, the increased coagulation and decreased platelets are impairing the cell’s ability to undergo mitophagy (Lee et al., 2016).
When platelets cannot undergo mitophagy, they undergo apoptosis, which leads to increased thrombus formation; this is especially true in diabetic patients who suffer from oxidative-stress destroying their mitochondria yet hindering mitophagy (Lee et al., 2016). COVID-19 patients suffer from hyper inflammation and iron buildup, both of which are stressful to platelets, and thus contribute to the decreased platelet count (Saleh et al., 2020).
Men have had more severe outcomes with COVID-19 than women. While the cause is unknown, it has been speculated that the TMPRSS2 receptor is involved (Singh et al., 2020). TMPRSS2 can be induced by androgen, but not estrogen, and localize to the mitochondria to regulate mitochondrial function (Singh et al., 2020). Older individuals have also had worse outcomes. Aging is accompanied by a decrease in mitochondrial function, which has been shown to worsen the severity of viral illness and is also linked to numerous age-related diseases.
reference link: https://www.frontiersin.org/articles/10.3389/fnagi.2020.614650/full
More information: Yuyang Lei et al, SARS-CoV-2 Spike Protein Impairs Endothelial Function via Downregulation of ACE 2, Circulation Research (2021). DOI: 10.1161/CIRCRESAHA.121.318902



{kind=link}