











 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Scientists using computer modeling to study SARS-CoV-2, the virus that caused the COVID-19 pandemic, have discovered the virus is most ideally adapted to infect human cells – rather than bat or pangolin cells, again raising questions of its origin.
In a paper published in the Nature journal Scientific Reports, Australian scientists describe how they used high-performance computer modeling of the form of the SARS-CoV-2 virus at the beginning of the pandemic to predict its ability to infect humans and a range of 12 domestic and exotic animals.
Their work aimed to help identify any intermediate animal vector that may have played a role in transmitting a bat virus to humans, and to understand any risk posed by the susceptibilities of companion animals such as cats and dogs, and commercial animals like cows, sheep, pigs and horses.
The scientists, from Flinders University and La Trobe University, used genomic data from the 12 animal species to painstakingly build computer models of the key ACE2 protein receptors for each species. These models were then used to calculate the strength of binding of the SARS-CoV-2 spike protein to each species’ ACE2 receptor.
Surprisingly, the results showed that SARS-CoV-2 bound to ACE2 on human cells more tightly than any of the tested animal species, including bats and pangolins. If one of the animal species tested was the origin, it would normally be expected to show the highest binding to the virus.
“Humans showed the strongest spike binding, consistent with the high susceptibility to the virus, but very surprising if an animal was the initial source of the infection in humans,” says La Trobe University Professor David Winkler.
The findings, originally released on the ArXiv preprint server, have now been peer reviewed and published in Scientific Reports.
“The computer modeling found the virus’s ability to bind to the bat ACE2 protein was poor relative to its ability to bind human cells. This argues against the virus being transmitted directly from bats to humans. Hence, if the virus has a natural source, it could only have come to humans via an intermediary species which has yet to be found,” says Flinders affiliated Professor Nikolai Petrovsky.
The team’s computer modeling shows the SARS-CoV-2 virus also bound relatively strongly to ACE2 from pangolins, a rare exotic ant-eater found in some parts of South-East Asia with occasional instances of use as food or traditional medicines. Professor Winkler says pangolins showed the highest spike binding energy of all the animals the study looked at – significantly higher than bats, monkeys and snakes.
“While it was incorrectly suggested early in the pandemic by some scientists that they had found SARS-CoV-2 in pangolins, this was due to a misunderstanding and this claim was rapidly retracted as the pangolin coronavirus they described had less than 90% genetic similarity to SARS-CoV-2 and hence could not be its ancestor,” Professor Petrovsky says.
This study and others have shown, however, that the specific part of the pangolin coronavirus spike protein that binds ACE2 was almost identical to that of the SARS-CoV-2 spike protein.
“This sharing of the almost identical spike protein almost certainly explains why SARS-CoV-2 binds so well to pangolin ACE2. Pangolin and SARS-CoV-2 spike proteins may have evolved similarities through a process of convergent evolution, genetic recombination between viruses, or through genetic engineering, with no current way to distinguish between these possibilities,” Professor Petrovsky says.
“Overall, putting aside the intriguing pangolin ACE2 results, our study showed that the COVID-19 virus was very well adapted to infect humans.”
“We also deduced that some domesticated animals like cats, dogs and cows are likely to be susceptible to SARS-CoV-2 infection too,” Professor Winkler adds.
The extremely important and open question of how the virus came to infect humans has two main explanations currently. The virus may have passed to humans from bats through an intermediary animal yet to be found (zoonotic origin), but it cannot yet be excluded that it was released accidently from a virology lab. A thorough scientific, evidence-based investigation is needed to determine which of these explanations is correct.
How and where the SARS-CoV-2 virus adapted to become such an effective human pathogen remains a mystery, the researchers conclude, adding that finding the origins of the disease will help efforts to protect humanity against future coronavirus pandemics.
Coronaviruses have a history of causing zoonotic outbreaks that occasionally achieve human-to-human transmission, giving rise to epidemics or pandemics as previously observed with Severe Acute Respiratory Syndrome Coronavirus (SARS) and Middle Eastern Respiratory Syndrome (MERS) coronaviruses [1,2,3]. While MERS and SARS remained limited to relatively smaller populations, Coronavirus Disease 2019 (COVID-19) caused by Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) has affected virtually the whole world, wreaking havoc on healthcare systems and costing millions of lives.
In December 2019, a SARS-like disease with unknown etiology was initially reported in Wuhan, China, the original epicenter of the pandemic. Later on, a single-stranded RNA virus from patients with severe respiratory illness, the novel betacoronavirus SARS-CoV-2 was determined as the infectious agent [4].
The novel coronavirus SARS-CoV-2, was shown to be capable of human-to-human transmission, leading the whole world to take action, lockdowns being imposed, followed by the World Health Organization (WHO) declaring the outbreak a pandemic [5].
Scientific community demonstrated rigor in investigating the virus itself and its pathogenesis once its genome sequence was revealed. It was quickly shown that the SARS-CoV-2 RNA genome is approximately 30,000 nt in length, including both coding and non-coding regions. The two thirds of its genome encodes non-structural proteins that aid in genome replication and RNA synthesis on the 5′ side.
The remainder of its genome codes for several structural proteins including Envelope (E), Spike (S), Nucleocapsid (N) and Membrane (M). Structural proteins help form the virion as well as functioning in various cellular processes [6]. S protein has been widely studied as it is responsible for forming the homotrimeric spike protein that binds to Angiotensin Converting Enzyme 2 (ACE2) promoting virus internalization [7, 8]. Proteolytic cleavage of the S protein by TMPRSS2 is also an important determinant of efficient viral and host cell membrane fusion that ultimately helps viral internalization [9].
RNA viruses are notorious for high mutation rates due in part to the error prone and fast nature of RNA dependent RNA polymerase [10]. Despite the RNA proofreading ability of coronaviruses [11], immune pressure and high mutation rate can eventually lead to protein sequence and structure changes with potential phenotypic impact, creating novel virus variants.
This has so far been observed in various instances, namely the early D614G variant, the Danish ‘Cluster 5’ variant, UK (B.1.1.7), South African (B.1.351) and Brazilian (P.1) variants have emerged during the first year of COVID-19 pandemic [12,13,14,15,16] (Table 1). N501Y amino acid change is highlighted as it is at the spike receptor binding domain (RBD), common to several variants, including UK and South African variants, where K417N and E484K substitutions are specific to the South African B.1.351 variant at the ACE2 interface (Fig. 1).
These variants are thought to differentially impact ACE2 binding [17] and are thought to potentially have an effect on transmission, effectivity of vaccines and disease progression. Several other variants were reported from the US including, ‘Columbus, Ohio variant’ which contains the N501Y and Q677H substitutions [18], as well as the B.1.429 variant containing L452R substitution that quickly dominated the viral pool in California outbreaks [19] and the fast spreading New York variant [20]. Impact of these and other variants on vaccine efficacy, virulence and pathogenesis is under investigation.
Table 1 Specifications of current SARS-CoV-2 variants and their transmission/neutralization capacities
| Current SARS-CoV-2 variant lineage nomenclature | Origin of detection | Protein changes of note | Neutralization by natural or vaccine ınduced antibodies | Effect on transmission | References |
|---|---|---|---|---|---|
| D614G | Europe | D614G | Increased | Increased | [95,96,97] |
| B.1.1.7 | UK | N501Y, ΔH69–V70, P681H | Decreased | Increased | [39, 98] |
| ‘Cluster 5’ | Denmark | ΔH69–V70 Y453F | Likely decreased | Likely eliminated | [99, 100] |
| B.1.351 | South Africa | N501Y, K417N, E484K | Decreased | Increased | [38, 101] |
| P.1 | Brazil | E484K, N501Y | Decreased | Increased | [39, 98] |
| B.1.427/B.1.429 | California, USA | S13I, W152C, L452R | Decreased | Increased | [46, 47] |
| B.1.526 | New York, USA | E484K or S447N and D614G, A701V, D253G | Slightly decreased for E484K, no change for S447N | Likely increased | [20, 48] |
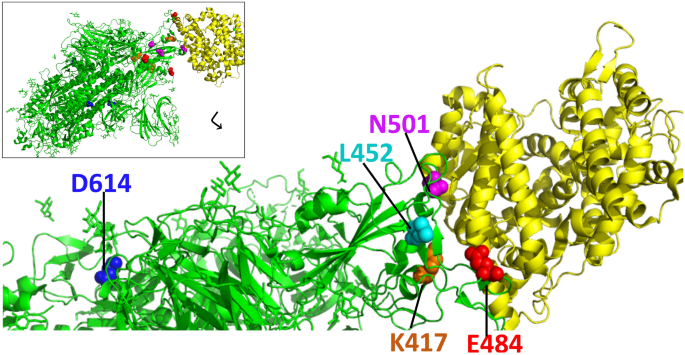
Extensive scientific research which accompanied the COVID-19 pandemic led to fast production of both antigen and nucleic acid based diagnostic tests and several vaccines in less than a year which was accompanied, but not yet overshadowed, by the emergence of SARS-CoV-2 variants. This review will highlight the impact of variants on disease management strategies, pathogenesis and vaccines.
SARS-CoV-2 Variants and Pathogenesis
How variants will impact SARS-CoV-2 pathogenesis is another topic of concern. SARS-CoV-2 has diverse tissue tropism. Due to its higher ACE2 affinity, it can efficiently infect the upper respiratory tract, such as the nasopharynx (also explaining its high transmissibility, along with asymptomatic transmission) unlike SARS-CoV [56].
Following the nasopharynx, it can enter the host via alveolar cells, vascular endothelial cells, alveolar macrophages and vascular endothelial cells. Mammalian gastrointestinal tract is also permissive to SARS-CoV-2 infection [78]. In addition to the classic respiratory symptoms, hyperinflammatory syndrome, gastrointestinal disease, coagulopathies, cardiac pathologies, and neurological problems associated with COVID-19 prompted detailed investigations [79,80,81].
Autopsy studies aiming at understanding the neurological symptoms, revealed cases of colocalization of SARS-CoV spike protein with neural/neuronal cells in olfactory mucosa of a subset of individuals deceased due to COVID-19. SARS-CoV-2 presence in CNS of a subset of deceased individuals was also detected both via staining and nucleic acid detection methods. These results indicate SARS-CoV-2’s ability in crossing the neural-mucosal interface in olfactory mucosa and entering the nervous system, providing an explanation toward some of the observed neurological symptoms [82].
Considering that novel variants have been causing structural changes in the spike protein, the changes in binding affinity of a variant spike to ACE2 or any processing change by TMPRSS2 or other downstream intracellular processes may affect SARS-CoV-2 variants’ tissue tropism or could change the course of infection. Future work will need to delineate if differential antibody recognition or ACE2 affinity exerted by the variants will be involved in changing viral pathogenesis.
What Determines Disease Severity, Impact of Biological Sex-Specific Traits and How the Variants May Affect It
The large number of people infected with SARS-CoV-2 during COVID-19 pandemic has revealed various outcomes of disease in a relatively short time.
For instance, while some people developed no symptoms, others suffered serious, even fatal disease. While a number of people got better in a short time, others, termed ‘long haulers’ or ‘long term COVID patients’ suffered symptoms lasting for many months. In some people, infection led to severe neurological symptoms such as stroke or encephalopathy, whereas other had none or mild neurological symptoms like a state of confusion, loss of taste or smell [82,83,84].
Over time, gender and age related differences in disease progression became obvious. People over 65 had worse outcomes compared to younger patients and women fared better compared to men [85, 86]. Knowledge on these details became obvious, mainly because of the variety of scientific research carried out in the world on COVID-19, due to the urgency of the situation and the large number of people who got the infection.
Research has focused on how biological sex impacts COVID-19 severity. Initial results highlighted the higher incidence of severe disease in males compared to females. Several mouse studies indicated a higher expression and activity for ACE2 protein in males [87]. In addition to ACE2, TMPRSS2, the serine protease used to cleave the spike protein to aid in viral internalization by allowing fusion of viral and host membranes, was also reported to have a higher expression in males due to higher androgen levels [88].
Furthermore, males may suffer elevated inflammatory immune responses compared to women, leading to fatal lung macrophage-monocyte infiltration and cytokine storm. Previous studies reported a higher C-reactive protein level as well as higher neutrophil to lymphocyte ratio in males, indicating an elevated inflammatory immune response and worse prognosis [86].
Another determinant of disease severity is associated with the fucosylation profiles of IgG antibodies against SARS-CoV-2. Level of afucosylated IgG antibodies show correlation with disease severity such that patients suffering severe COVID-19 have high levels of afucosylated IgG compared to those with mild symptoms. This increased afucosylated IgG profile amplifies pro-inflammatory cytokine response, leading to acute case manifestation [89].
Interestingly, the level of afucosylated anti-spike RBD IgG was elevated in hospitalized males compared to females [90]. Antibody response elicited in the presence of different SARS-CoV-2 variants in both sexes will be a strong determinant of disease progression. In the light of this information, investigational tools may be adapted to understand the fucosylation levels of therapeutic antibodies. Furthermore, convalescent plasma therapy strategies can be manipulated to include volunteers with high levels of fucosylated IgG. These factors will be particularly important especially if certain variants are indeed causing worse prognosis or if variants give rise to differing antibody or immune profiles.
In addition to IgG fucosylation levels, there are several other variables that affect the success rate of convalescent therapy [91]. Location is thought to be one of the factors contributing to the effectivity of convalescent therapy, as different variants tend to circulate in specific areas. Sera obtained from an individual in a certain area is presumed to have generated an antibody response against a particular variant prevalent in that area, therefore will be most useful for a patient infected with the same variant.
Being hospitalized with severe COVID-19, being older and having male sex are associated with having a higher antibody response against SARS-CoV-2, making these individuals better candidates as plasma donors [91]. While males have higher plasma antibody titers, they also have higher levels of afucosylated IgG. Considering the importance of plasma donor in relation to the variants as well as the sex-specific antibody profiles, the selection criteria for convalescent therapy should include sex-specific variations, location and the prevalence of variants.
Not only the SARS-CoV-2 spike variants but also the variants of ACE2 receptor were demonstrated to determine the affinity of host-virus interactions [92, 93]. For instance, ACE2 K31R and E37K substitutions showed decreased affinity where K26R and T92I substitutions showed increased affinity for wild type SARS-CoV-2 spike protein [93].
Rare X-linked alleles, including the ones encoding E37K, are observed twice as frequently in females, suggesting that these missense variants may predominantly affect SARS-CoV-2 affinity in females [94]. Modeling studies coupled with biochemical interaction/affinity evaluation of ACE2 substitutions with spike variants are likely to provide insight toward the potential gender-specific impact of spike variants in host–pathogen interactions as well as susceptibility to SARS-CoV-2 variants.
Conclusions
More variants are expected to emerge especially at the spike RBD as it is under immune selection due to being a major epitope for neutralizing antibodies. Therefore, more variants with differing antibody recognition and ACE2 affinities are expected. Future studies will need to delineate the immune response and pathogenesis associated with novel SARS-CoV-2 variants. Considering varying antibody recognition and neutralization states for different variants, different downstream immune responses can be expected that could eventually affect disease outcome.
Overall, novel SARS-CoV-2 variants run the risk of changing immune response elicited by the host, vaccine and therapeutic antibody efficacy, disease pathogenesis and prognosis. As variants tend to accumulate at the spike protein’s RBD, ACE2 affinity or antibody binding changes can lead to different pathogenesis and immune responses.
Monoclonal antibody therapeutics and convalescent plasma therapy options should also be adaptable in the light of current or potential novel variants. Scientific advances achieved by quick and successful vaccine design should now be implemented to keep the vaccines as effective against the current and potentially upcoming SARS-CoV-2 variants.
reference link: https://link.springer.com/article/10.1007/s12033-021-00353-4
More information: Sakshi Piplani et al, In silico comparison of SARS-CoV-2 spike protein-ACE2 binding affinities across species and implications for virus origin, Scientific Reports (2021). DOI: 10.1038/s41598-021-92388-5



{kind=link}