









 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Despite the slow evolutionary rate of SARS-CoV-2 relative to other RNA viruses, its massive and rapid transmission during the COVID-19 pandemic has enabled it to acquire significant genetic diversity since it first entered the human population.
This led to the emergence of numerous variants, some of them recently being labeled “variants of concern” (VOC), due to their potential impact on transmission, morbidity/mortality, and the evasion of neutralization by antibodies elicited by infection, vaccination, or therapeutic application.
The potential to evade neutralization is the result of diversity of the target epitopes generated by the accumulation of mutations in the spike protein. While three globally recognized VOCs (Alpha or B.1.1.7, Beta or B.1.351, and Gamma or P.1) remain sensitive to neutralization albeit at reduced levels by the sera of convalescent individuals and recipients of several anti-COVID19 vaccines, the effect of spike variability is much more evident on the neutralization capacity of monoclonal antibodies.
The newly recognized VOC Delta or lineage B.1.617.2, as well as locally accepted VOCs (Epsilon or B.1.427/29-US and B1.1.7 with the E484K-UK) are indicating the necessity of close monitoring of new variants on a global level. The VOCs characteristics, their mutational patterns, and the role mutations play in immune evasion are summarized in this review.
Organization of SARS-CoV-2 Genome and Spike Protein
SARS-CoV-2 belongs to order Nidovirales, family Coronaviridae, subfamily Orthocoronavirinae, and genus Betacoronavirus. Virions of coronaviruses are spherical with average diameters of 80 to 120 nm. They are enveloped with positive single-stranded (ss) RNA genomes. The genomic analysis of three newly discovered coronaviruses showed that SARS-CoV-2 has 79% and 50% sequence similarity with SARS-CoV and MERS-CoV, respectively [8,9].
The coronavirus with the most similar genome to SARS-CoV-2 is horse-shoe bat virus RaTG13 Rhinolophus affinis with 96% of similarity [8,10]
SARS-CoV-2 genome is in the form of ssRNA with positive sense and a length of approximately 30,000 nucleotides. This non-segmented genome includes a 5′-untranslated region (UTR), followed by replicase complex (ORF1a and ORF1ab), structural genes for spike (S), envelope (E), membrane (M), nucleocapsid (N) proteins, and several open reading frames (ORFs) for accessory proteins inserted between four structural genes, ending with 3′-UTR with poly A tail [1,11] (Figure 1).
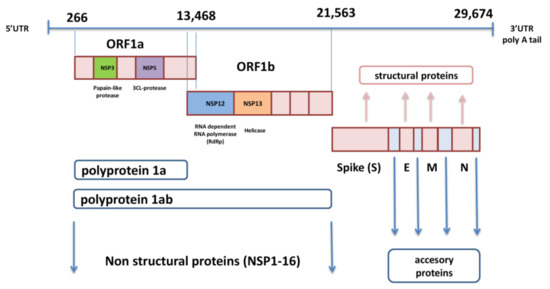
The ORF1a and ORF1b genes, located next to each other near 5′-UTR, occupy two-thirds of SARS-CoV-2 genome and encode polyproteins pp1a and pp1ab. These two polyproteins are cleaved with autoproteolytic enzyme into 16 non-structural proteins (nsp1-16) that are involved in viral replication, transcription, immunomodulation, gene transactivation, and resistance to innate antiviral response [12]. The last third of the genome contains genes for structural and accessory proteins.
The S gene encodes spike glycoprotein, which is the most prominent protein of the virion and enables viral entry into the target cell [13]. The M glycoprotein contains three domains, C terminal-, transmembrane-, and N terminal-domain, and it is necessary for the assembly and budding of virions [14].
The envelope protein also includes three domains and plays an important role in the pathogenesis of COVID-19 infection because its C-terminal domain binds to human tight junction protein PALS1 [15]. The nucleocapsid binds to viral RNA and influences the replication performance of SARS-CoV-2 [16]. Accessory proteins significantly contribute to evasion of the innate immune response by meddling with interferon (IFN) synthesis and obstructing signal pathways within the cell [17].
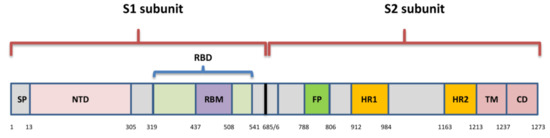
The spike is a transmembrane glycoprotein that is 1273 amino acids long and in the shape of a homotrimer. It comprises the receptor binding domain (RBD) that interacts with host cell receptor angiotensin converting enzyme 2 (ACE2) [18]. The SARS-Cov-2 S protein shares amino acid sequence similarity of 76.7–77% with SARS-CoVs from humans and civets, 75–98% with bat coronaviruses of the same subgenus (Sarbecovirus), and 90.7–92.6% with pangolin coronaviruses [8].
Spike includes two subunits: S1 (aa 1–685) and S2 (aa 686–1273) (Figure 2). The S1 subunit comprises an N-terminal domain (NTD) and RBD (aa 319–541), while the S2 subunit is composed of a fusion peptide (FP) (also called S2′ subunit), heptapeptide domain 1 and 2 (HPD1, HPD2), transmembrane domain (TM), and cytoplasm domain (CD) [19]. The RBD is the key player within the S1 subunit for the attachment of SARS-CoV-2 to ACE2 [20] and is therefore a very important target for antiviral drugs and antibodies [21].
It contains a core structure and receptor binding motif (RBM) (aa 437–508), which is the most variable part of spike protein that is important for binding to the outer surface of ACE2 [18]. There is only 73% similarity between RBD of SARS-CoV-2 and SARS-CoV, although they both bind to human ACE2 [22].
The characterization of the mechanical stability of the RBD of SARS-CoV-2 has shown it to be stiffer compared to SARS-CoV, which has important consequences in binding to ACE2, as it can withstand Brownian and cellular forces while remaining in close contact during the initial steps of cell entry [23]. The S protein persists in the open and closed form [24].
In the closed form, recognition motifs are hidden, and in the open form, the so-called receptor-accessible state, RBD with RBM are in up conformation. While in the up conformation, they stick out from the homotrimers and enable the binding to the receptor, fusion process, and virus entry into the host cell [20]. The fusion of two membranes is the crucial step in the viral life cycle [25].
It was suggested that certain key residues are crucial for stabilization of the spike protein during transitions from close to open conformations prior to ACE2 recognition [26]. Several high-frequency contacts are formed between the NTD and RBD that are responsible for the local conformational stability and play a role during the transition from closed to open state.
The S2 subunit has three conformational states: (1) pre-fusion native state; (2) pre-hairpin intermediate state; and (3) post-fusion hairpin state. The S1 and S2 subunits, which are non-covalently bound in the pre-fusion state, have a united role in binding to the receptor of the host cell [22].
The S1 subunit interacts with the receptor and enables attachment of the virion [27], while the S2 subunit is involved in the fusion process. The S protein is cleaved by host proteases, type II transmembrane serine proteases (TMPRSS2), and furin, at the junction of S1 and S2 at a polybasic cleavage site (S2′) [28,29]. The cleavage at the S2′ site activates the proteins, which induce irreversible conformational changes in the S protein, which is crucial for the fusion of viral and cell membranes [24].
The insertion of four amino acids (PRRA) at the polybasic cleavage site represents a specific genomic characteristic of SARS-CoV-2 [28]. This site is not observed in related coronaviruses except in a bat-derived coronavirus from Rhinolophus malayanus (RmYN02), which has an insertion of three amino acids (PAA) [30]. Some studies indicate that this furin-cleavage site induces the instability of SARS-CoV-2, causing conformational changes that are needed for the binding of RBD to the receptor [31].
Viruses, such as SARS-CoV-2, besides innate immunity, induce both humoral and cellular adaptive immune response, triggering different defense mechanisms in order to fight acute infection. NK cells, monocytes (macrophages), and IFN type I are crucial in the response to this virus. A fall in the number of NK cells [32] and plasmacytoid dendritic cells (main source of IFN type I) and dominating interleukin (IL)-6 producing monocytes are characteristic of inappropriate SARS-CoV-2 innate immune response [33]. https://pubmed.ncbi.nlm.nih.gov/33357409/Large (19 June 2021) trials, such as RECOVERY, established that anti-IL-6 in combination with steroids is a potential option for hypoxic patients with evidence of hyperinflammation [34].
There are data showing that a statistically significant correlation exists for some common variants in three genes linked to the innate immunity, MBL2, TMPRSS2, and CD27. MBL2 encodes a mannose-binding protein C that binds to mannose, activating the lectin complement pathway; TMPRSS2 cleaves the spike protein and ensures viral internalization [35], while the CD27 receptor is required for the generation and long-term maintenance of T-cell immunity.
There are some data suggesting that trained innate immunity might also have a role in the protection against COVID-19 [36,37]. Several clinical trials are investigating whether unrelated vaccines, such as the measles, mumps, rubella vaccine, and the BCG vaccine can provoke trained innate immunity and improve protection against COVID-19 [38].
It is important to stress that recovery from COVID-19 infection is linked to appropriate immune response and disease severity is correlated to the impaired immune reaction. It is well known that this virus with its particular potential to inactivate the IFN-based response leads to the weakening of innate immunity.
In addition, once present in the host cell, the SARS-CoV-2 activates the NOD-like receptor family, inducing the formation of an inflammasome. This contributes to the release of the pro-inflammatory cytokines, IL-1, IL-6, TNF, and IL-18. The NF-ĸB pathway is activated after interaction of the viral RNA with Toll-like receptors and enhances pro-inflammatory cytokines production. Thus, the inflammation starts and leads to the release of a number of cytokines from activated immune cells and the so-called cytokine storm, which can be life-threatening, happens [39].
Humoral immune response to SARS-CoV-2 is mediated by antibodies specific mainly to the spike glycoprotein, all of its parts including NTD, and the nucleocapsid protein [40]. These antibodies neutralize viral binding to cells expressing ACE2 receptors and infection of these cells [41]. Many studies that examine the duration of protection by functional neutralizing antibodies and the potential for re-infection have shown that most patients with COVID-19 have virus-specific IgM, IgA, and IgG responses in the days after infection [42].
In individuals with mild COVID-19, a rapid decline of RBD-specific IgG titers within 2–4 months has been observed in several studies, suggesting that SARS-CoV-2-induced humoral immunity might not be long-lasting in individuals with mild disease [43]. Antibody titers were significantly higher in patients with severe disease than in patients with mild disease and were associated with clinical outcomes [44].
However, a comprehensive study of adaptive immunity to SARS-CoV-2, which also examined the association with disease severity, showed that the concentration of neutralizing antibodies was not correlated with COVID-19 severity [45]. There is no pre-existing immunity to SARS-CoV-2 in the population, except through cross-reactivity with other coronaviruses [46]. It is also important to evaluate memory B-cells in addition to antibody measurement to better characterize humoral immunity. Although high circulating titers of neutralizing antibodies are common surrogates of protective immunity, there are many situations when circulating antibodies do not reach sufficient levels, and additional input from memory B-cells is necessary [47].
If circulating antibodies disappear over time, data suggest that robust memory B-cells are likely to provide a quick source of protective antibody in the case of potential SARS-CoV-2 re-infection. In addition, in infection with variants that can partially escape neutralization by present circulating antibodies [48,49,50], one will need vital memory B-cells to re-enter germinal centers and transform in order to respond to novel spike epitopes [51].
In addition, human monoclonal antibodies (mAbs) targeting both the NTD and RBD of SARS-CoV-2 have been isolated, with those targeting RBD being especially potent. These antibodies are used clinically [52,53], in therapeutic and prophylactic modes. Moreover, the selection of antibody mixtures with non-overlapping escape mutations should help and prolong the effectiveness of antibody therapies in SARS-CoV-2 infection [54].
Following the infection, a certain number of HLA-DR+ T-cells, both CD4+ and CD8+, rises in the first 7–10 days after the first symptoms and declines after three weeks [55,56,57]. The CD4+ T-cell response to SARS-CoV-2 predominantly consists of T-helper-1 (Th1) cells, which are characterized by high IFN-γ secretion and specificity for the structural spike glycoprotein, the membrane protein, and the nucleocapsid protein. CD8+ T-cell response specific to SARS-CoV-2 also produced IFN-γ and tumor necrosis factor (TNF).
SARS-CoV-2-specific T-cells express perforin and granzymes after in vitro reactivation with viral antigens. It was also shown that during the convalescent phase, T-cells had a memory phenotype, both CD4+ and CD8+ T-cells expressing IFN-γ, IL-2, and TNF [58]. Response from T follicular helper (Tfh) cells is crucial to the development of strong humoral immunity through the formation of germinal centers and provision of co-stimulation (CD40–CD40-L interaction and cytokines) to B-cells [59].
A single-cell RNA sequencing study of the CD4+ T-cells specific to SARS-CoV-2 found an increased proportion of Tfh cells in patients with severe disease [60]. Other risk factors for severe COVID-19 are increased numbers of Th17 cells, T-cells expressing exhaustion markers (such as PD-1), and the depletion of both αβ and γδ T-cells [61]. The recognition of SARS-CoV-2 antigens by pre-existing and cross-reactive T-cells created during previous infection with human coronaviruses is also possible [62].
In the recent study [63], the authors have suggested that T-cell response and the binding of antibodies to the spike protein induce early protection in COVID-19. After mRNA vaccines, all individuals develop spike-specific T-cells, while 80% develop spike-binding antibodies 10 days after the first dose. They are suggesting that a lack of neutralizing antibodies is not essential to prevent against COVID-19. With the exception of killed whole-virus vaccines, all current vaccines offer S protein as the target immunogen, limiting T-cell immunity to spike epitopes. For the T-cell epitopes, a population exposure analysis proposed a set of epitopes that is estimated to provide broad coverage worldwide [64].
Genetic Variability of SARS-CoV-2 and Classification of Variants
The genetic diversity of SARS-CoV-2 is the result of errors generated by its RNA-dependent RNA polymerase (RdRp) and recombination [65]. The capacity of coronaviruses to recombine is associated with the strand switching ability of RdRp, and it is likely that it played a significant role in their evolution. Although coronaviruses have a slower mutation rate relative to other RNA viruses because of their proofreading 3′-to-5′ exoribonuclease (nsp14), the consequences of accumulating mutations are still a major concern. It became obvious that the accumulation of amino acid mutations might affect the transmissibility of the virus, its cell tropism, and its pathogenicity, presenting a serious challenge for the efficiency of current vaccines and diagnostic assays.
Until recently, the observed diversity among SARS-CoV-2 sequences has been low. The earliest spike protein mutation D614G of SARS-CoV-2 in Europe was identified in January 2020 in Germany [66]. Since then, the strain harboring D614G has become the dominant pandemic variant in most countries, possibly because the mutation enabled a relative fitness advantage to the original Wuhan strain and enhanced infectivity.
The accumulating number of SARS-CoV-2 variants has developed a need for their classification into groups such as lineages and clades. On 31 May 2021, the World Health Organization (WHO) introduced names based upon the Greek alphabet for important variants in order to simplify public communication around variants and enable referring to variants in a geographically neutral fashion [67]. However, this does not replace the three current nomenclature systems: GISAID (Global Initiative on Sharing All Influenza Data), Nextstrain, and PANGO.
There are 8 clades of SARS-CoV-2 or hCoV-19 (S, O, L, V, G, GH, GR, and GV) identified by the GISAID database [68], 11 major clades (19A, 19B, and 20A–20I) recognized by Nextstrain, while Rambaut et al. [69] and the software team of the Phylogenetic Assignment of Named Global Outbreak Lineages (PANGOLIN) proposed 6 major lineages (A, B, B.1, B.1.1, B.1.177, B.1.1.7) now known as PANGO nomenclature.
If a new, emerging variant possesses specific genetic markers that have been associated with increased transmissibility, morbidity and mortality, and ability to evade natural immunity as well as reduced neutralization by therapeutic antibodies or vaccination, reduced efficacy of treatments or potential diagnostic impact, it may be labeled “variant under investigation (VUI)” or “variant of interest (VOI)”, and if its prevalence and expansion surpasses the national level, it can be marked “variant of concern (VOC)”. If there is evidence that a variant has developed features that significantly reduce the effectiveness of existing prevention or intervention measures, it can be termed a “variant of high consequence” [70,71,72].
So far, there are four globally recognized variants of concern: Alpha or lineage B.1.1.7 (UK), Beta or lineage B.1.351 (South Africa), Gamma or lineage P.1 (Japan/Brazil), and Delta or lineage B.1.617.2 (India) [70,71,72,73]. Another was acknowledged as VOC in the UK and by ECDC—B.1.1.7 with E484K and two others by the US—Epsilon or B.1.427/29 [71,72,73].
Alpha or lineage B.1.1.7 (also known as 20I/501Y.V1 and VOC-202012/01) emerged in September 2020 in Southeastern England. It harbors seven missense mutations and three deleted residues in the spike protein [74]. Due to its enhanced transmissibility, it quickly spread worldwide and it is reported, as of 1 June 2021, in 160 countries. In February 2021, Public Health England (PHE) recognized B.1.1.7 with E484K mutation as a new VOC (VOC-202102/02), and it has since been identified in the US. However, this variant has not been detected in the UK since March 2021 but is continuing to spread outside the UK based on sequence data.
Beta or lineage B.1.351 (also known as 20H/501Y.V2 variant) was first detected in the Eastern Cape province of South Africa in late 2020 [75]. It contains seven mutations and three deleted residues in spike protein. This variant is of the greatest concern in regard to immune escape for its three mutations within RBD and has since been spread to 113 countries. Variant Gamma or P.1 (also known as 20J/501Y.V3 variant) arising from lineage B.1.1.28 was first described in Brazil and Japan in December 2020 and later classified as VOC due to 11 spike mutations, including the same three in RBD as South African variant [76].
It has since been reported in 64 countries. Lineages B.1.429, defined by four and B.1.427 by two spike mutations are recognized as VOC in the US and as VOI Epsilon by WHO [77]. They were first identified in California (both also known as CAL.20C and 20C/S:452R), where they reached prevalence of more than 50% as of February 2021. As of June 2021, more than 60 countries reported cases caused by a newly recognized variant—lineage B.1.617 (also known as G/452R.V3) and its three sublineages, the first two detected in December 2020 and the third detected in February 2021 in India [70].
However, it has since become evident that only sublineage B.1.617.2 is associated with greater public health risk, which is why it is now the only sublineage of B.1.617 that is recognized as VOC—Delta [67]. Sublineage B.1.617.1 has been reclassified to a VOI (variant Kappa), and while it is still demonstrating increased transmissibility, global prevalence appears to be declining. Based upon reports, the prevalence of B.1.617.3 is low, and it is no longer classified as either a VOC or VOI.
The main speculation about the origin of novel variants with accumulated mutations is proposing that they evolved within immunosuppressed chronically infected patients who supported high viral replication for months and may have been treated with immune plasma or monoclonal antibodies [78,79,80]. However, since the lineages usually contain circulating intermediate mutants, the diversity within some lineages cannot be explained only by a single long-term infection in one individual [75].
Implications of SARS-CoV-2 Variants in Immune Evasion
Although different lineages are defined by mutations in more than one region of the genome, the most attention is paid to nonsynonymous changes in the S gene, which can alter the spike protein and influence its role in viral entry. This role of spike has determined it as an ideal target for immune response and also made it the primary target for most currently approved vaccines.
Amino acid changes have been observed across the entire spike protein, but the exact location defines the impact of each substitution. The NTD and RBD are the most diverse regions, and most mAbs against SARS-CoV-2 that have been characterized target the RBM, and some are specific for RBD core- and NTD as well [81]. Changes in spike residues within major epitopes may reduce or ablate antibody binding and neutralization, which would lead to the diminished efficacy of antibodies, derived by natural infection or vaccination.
However, changes are found to occur also within the conserved C-terminal domain of the S1 and the S2 subunit. These regions are important for conformational changes within S, which is needed for viral attachment and fusion, and may elicit still unknown neutralizing responses [82].
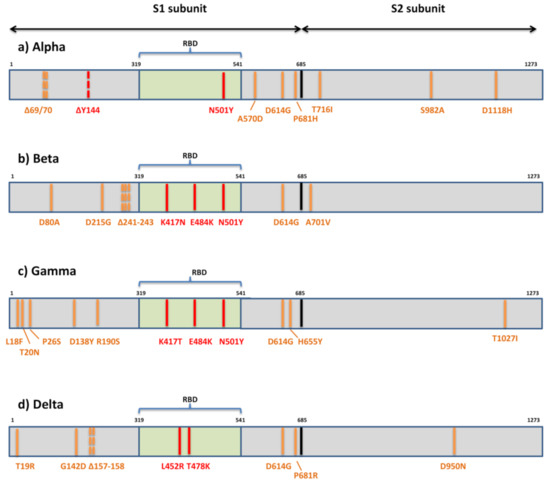
The first variant Alpha or B.1.1.7 that raised global concerns about increased transmissibility and potential immune evasion harbors seven missense mutations (N501Y, A570D, D614G, P681H, T716I, S982A, D1118H) and three deletions in spike (69/70del and 144del) (Figure 3).
The three deleted residues are located within NTD, only one mutation (N501Y) is within RBM, three are displayed in the C-terminal domain (CTD) of S1, and three are displayed within S2. Various studies have so far demonstrated the reduced potency of neutralizing antibodies against B.1.1.7 [49,83,84,85,86,87,88]. These studies share the general conclusion that variant B.1.1.7 remains sensitive to neutralization, though at moderately reduced levels, by sera of convalescent individuals and recipients of several anti-COVID19 vaccines.
The reduction in neutralization levels were on average 3-fold (ranging from 1.5-10-fold) for convalescent sera and ≈2-fold for sera of vaccine recipients (mRNA-, vector-, and subunit-based) [49,83,84,85,86]. However, when various mAbs were tested against this variant, it was uniformly shown that the B.1.1.7 variant can escape neutralization mediated by a fraction of RBM-specific antibodies and by most NTD-specific antibodies [49,83,88].
The proposed explanation for the more serious effect of spike mutations on neutralization by mAbs than by sera is the polyclonality of serum neutralization [84]. It is supported by the observation that a single mutation can diminish the binding of a single mAbs but not of other antibodies in the same binding cluster. A single mutation cannot affect all antibodies in the same cluster, since it seems that each antibody has well defined and unique molecular contact with the same specific epitope. Therefore, polyclonal sera are less susceptible to changes in neutralization due to a single mutation. Polyclonal sera also contain non-neutralizing antibodies whose role is yet to be elucidated.
The part of the diminished neutralizing effect of antibodies against the B.1.1.7 variant can be attributed to the only RBM mutation—N501Y. This mutation, shared by three globally recognized VOCs, is thought to be the result of viral adaptive evolution [89] and has been shown to increase affinity for ACE2 [85,90,91,92]. The enhanced binding affinity may be contributed to additional interactions with ACE2 that are allowed by 501 change—the new hydrogen bonds at residues 41 and 353 and also to a more open conformation of the RBD [93,94].
While some report its antigenic impact to be limited to a few mAbs with no significant effect on neutralization by convalescent or vaccinees sera [83], others show that the increase in transmission is combined with the reduction in the neutralization potency of convalescent sera [85]. The explanation for this lies not in the disrupted binding of antibodies to changed RBM but in competition of antibodies with ACE2 for binding to RBM. Thus, all changes in RBM that confer increased affinity for ACE2 will make the virus more difficult to neutralize.
The significant resistance of B.1.1.7 to neutralization by NTD-specific antibodies should be explained by the presence of three deleted residues in this region. The neutralization effect of these antibodies can be attributed to the role that NTD has in viral entry. While it was not yet determined for SARS-CoV-2, the NTD has a role in attachment to host cells in several CoV family members [95].
For SARS-CoV-2, it has been proposed that NTD interacts with auxiliary receptors in cell types that do not express ACE2 (e.g., DC-SIGN/L-SIGN) [96]. The NTD deletion H69/V70 is observed in B.1.1.7. and B.1.298 (Danish mink) but has not been associated so far with escape from NTD-specific antibodies [88].
A combination of del H69/V70 and N501Y was shown to increase infectivity in vitro [97]. On the other hand, deletion Y144 has been found to abrogate binding to neutralizing antibodies [49,52,88,98]. It can still not be determined whether NDT mutations are the result of immune selection or are generated as part of viral fitness improvement.
Other spike mutations of B.1.1.7 belong to the C-terminal domain of S1 and S2 and were not so far perceived to affect antibody neutralization. However, mutations within these regions might affect the conformation of RBD, attachment, and fusion, requiring further studies to determine their consequences and possible indirect effect on immune evasion. The extensively studied D614G was found to increase the ability of RBD to shift to the up position, which is necessary for interaction with ACE2 [99].
This resulted in the increased infectivity and transmissibility observed for the D614G variant relative to the original SARS-CoV-2 strains [100]. The P681H change is adjacent to the furin cleavage site and could potentially have an effect on S1/S2 cleavage and therefore on cell entry and infectivity.
The variant of the greatest concern in regard to immune escape, Beta or B.1.351, contains seven mutations (D80A, D215G, K417N, E484K, N501Y, D614G, A701V) and three deletions (241/242/243del) in the spike protein [75] (Figure 3). Two mutations (D80A, D215G) and three deleted residues are in the N-terminal domain of S1, one (A701V) is in loop 2 of S2 and 3 are at key residues in the RBD (K417N, E484K, N501Y).
So far, there are multiple studies showing that B.1.351 decreases the neutralization capacity of antibodies elicited by infection with previous variants or vaccination [48,83,101,102,103,104,105]. This reduction in neutralizing potential for B.1.351 is most frequently detected in individuals with low antibody levels, and it is declining more rapidly with time [105], heightening concerns about re-infection or suboptimal protection by current vaccines.
The problem in the non-vaccinated population exists because most people infected with SARS-CoV-2 develop only low to moderate titers, while higher titers are only observed in severely ill hospitalized individuals. The loss of neutralizing activity of convalescent plasma against B.1.351 ranged from 11 to 33-fold and by sera of vaccinees from 3.4 to 8.5-fold [50,83,101,103,104,105,106]. In addition, the B.1.351 variant showed resistance to neutralization by most NTD-specific and a number of RBM-specific mAbs [83,103,107].
The resistance to antibody neutralization of the B.1.351 variant is mainly ascribed to three mutations within RBD (K417N, E484K, N501Y). N501Y probably does not impair neutralization on its own but rather in combination with other two, which were found to partially compromise neutralization generated by previous infection or vaccination [48,103,106,107,108]. The result of the change at position 417 is loss of the polar interaction with residue D30 on human ACE2 [82].
However, a combination of K417N and N501Y was shown to enhance the binding with ACE2 and reduce binding with antibodies [109]. This improvement in receptor binding is supported by the observation of this mutation in a virulent mouse adapted strain of SARS-CoV-2 [110]. K417N was shown to be crucial to viral escape, effectively abrogating neutralization by some of the most common and potent neutralizing antibodies to SARS-CoV-2 [103]. Contrary to this, others [107] indicate that it may contribute to neutralization by enhancing the probability of conversion to the open conformation of the S protein, thus exposing epitopes to antibody neutralization.
Mutation E484K, which emerged independently in over 50 lineages, also corresponds with improved binding to ACE2. It enhances the binding affinity of N501Y for ACE2 still further but has been associated with immune escape from both mAbs and polyclonal sera as well [48,49,83,106,107].
Its location is within the RBD binding cleft, and it is considered to be a dominant neutralizing epitope [75,108,111]. The residue 484 can mutate into a diversity of different amino acids (E484A, E484G, E448D, and E484K) under the pressure of SARS-CoV-2 convalescent sera and exhibits resistance [112]. It is believed that the impact of mutation 484 on immune evasion is significantly augmented by the presence of other two RBD mutations in this variant, but its impact as the single point mutation was demonstrated as well [106,112].
The B1.1.7 variant bearing the E484K mutation emerged and was recognized as a variant of concern in the UK and Europe, since it appears to be responsible for a significant additional loss of neutralization capacity of monoclonal and polyclonal antibodies [49].
Monoclonal antibodies were shown to lose almost 50% of neutralizing activity against B.1.1.7 carrying E484K. A combination of E484K with various NTD mutations (particularly deletions) might prove to be even more effective in immune evasion [113], which is of the most significance in cases of both Beta variant and B1.1.7 with E484K.
The third globally recognized VOC, Gamma or P.1, is carrying 11 spike mutations. Five mutations are located within NTD (L18F, T20N, P26S, D138Y, R190S), three in RBD (K417T, E484K, N501Y), two in the C-terminal domain of S1 and near the furin cleavage site (D614G, H655Y), and one in S2 (T1027I) (Figure 3). Convalescent and vaccinee sera show a significant loss of neutralizing activity against P.1, but the reduction is not as substantial as against B.1.315 [114,115,116].
The loss of neutralizing activity of convalescent plasma against P.1 ranged from 6.5 to 13-fold and by sera of vaccinees from 2.2 to 2.8-fold [114,115], meaning that the neutralization of P.1 was not as severely compromised as that of B.1.351 and only slightly weakened compared to that of B.1.1.7. Not surprisingly, the neutralization activity of mAbs against P.1 is reduced much in the same manner as in B.1.351, since triple RBD mutations are mostly the same in both variants [114].
The reason for the differences in neutralization of B.1.351 and P.1 by the immune serum presumably reflects the difference in the mutations introduced outside the RBD. The role of NTD-specific neutralizing antibodies is not nearly yet defined. It was thought that extensive N-linked glycan shielding of NTD is diminishing its antigenicity, but in vitro studies showed the significant neutralizing capacity of some NTD-specific antibodies [52].
The fact that NTD is under selective pressure of human immune response is supported by the identification of NTD deletions in immunocompromised hosts with prolonged infections [79]. It is possible that neutralization assays based on target cells over-expressing ACE2 receptors are responsible for underrating the role of NDT-specific antibodies. Since NTD changes are much more distinct among three major VOC, it seems likely that neutralization variation among them is rather due to differences in NTD than RBD.
In January 2021, the emergence of a novel variant in California carrying an L452R mutation in the RBD was reported [77]. This variant (Epsilon) comprises two separate lineages B.1.427 and B.1.429, the first carrying two spike mutations (L452R, D614G) and the second carrying four (S13I, W152C, L452R, D614G). It is assumed that they emerged as early as May 2020 and they gained VOC status in the US due to significant increase in frequency from September 2020 to January 2021. In February 2021, they were identified in >50% of all sequenced cases in California and many other states [117].
They were shown to display moderate resistance to neutralization by convalescent sera (4–6.7-fold) and sera of vaccine recipients (2–2.9-fold) [48,117]. The RBD mutation L452R, shared by these lineages, is not located in the part that directly interacts with ACE2, but it is speculated that it may cause structural changes in the region that promote the interaction between the spike protein and its ACE2 receptor.
Thus, the infectivity of pseudoviruses carrying L452R was shown to be higher than of the D614G variant but slightly reduced compared to that of N501Y variants [117]. The similar mechanism of RBD structural change due to L452R is offered in explanation of the reduced neutralization capacity of antibodies. This mutation, among several other RBD mutations, was selected by a panel of antibodies in vitro [112].
The emerging variant B.1.617 comprises three distinct sublineages (B.1.617.1, B.1.617.2, B.1.617.3) with different mutational profiles [70]. However, only sublineage B.1.617.2 or Delta is now internationally recognized as VOC. It is characterized by spike mutations T19R, G142D, Δ157-158, L452R, T478K, D614G, P681R, and D950N (Figure 3). The other two sublineages have a similar mutational profile: B.1.617.1 is defined by the spike amino acid changes G142D, E154K, L452R, E484Q, D614G, P681R, and Q1071H, and B.1.617.3 is defined by T19R, L452R, E484Q, D614G, P681R, and D950N. The presence of RBD mutations L452R, E484Q, and D614G in the C-terminal domain of S1 may result in the higher transmissibility of these sublineages due to their known impact on ACE2 binding and conformational changes important for ACE2 binding.
All three sublineages of B.1.617 display P681R adjacent to the furin cleavage site and have enhanced S cleavage by furin, which is hypothesized to be enhancing transmissibility and pathogenicity [118]. Although the sublineage B.1.617.2 was initially considered to be as transmissible as B.1.1.7 [119], further evidence from the UK, based on the likelihood that close contacts of a person infected with the Delta variant will themselves become infected—the “secondary attack rate”, suggest that this variant may be over 60% more transmissible than the Alpha variant [120].
By recent report, more than 90% of new COVID-19 cases in the UK involve the Delta variant. The spread of the Delta variant is also registered in the US, where it now accounts for more than 6% of all infections (more than 18% of cases in some Western U.S. states) [121].
The impact on the immune escape capacity of three sublineages of B.1.617 is expected, owing to RBD mutations L452R, T478K, and E484Q and their combination with NTD mutations and deletions, particularly in the case of B.1.617.2. A similar change at position 478 (T478I) was previously selected in vitro and shown to exhibit reduced neutralization by monoclonal antibodies and human convalescent sera [112].
One of the first studies on B1.617.1 revealed that the neutralization capacity of convalescent sera and sera of recipients of inactivated killed vaccine was retained [122]. Other studies have reported a moderate reduction in neutralization of B1.617.1 by the sera of convalescents and recipients of mRNA vaccines and resistance to monoclonal antibodies approved for COVID-19 treatment [123,124,125]. The E484Q was found to have slightly milder impact but still corresponding to the effect of E484K, which is 10-fold reduction in the neutralization by sera of vaccine recipients. In addition, the combination of L452R and E484Q was not shown to have an additive effect; rather, the loss of sensitivity was similar to that observed with each mutation individually [124].
Finally, the impact of emerging SARS-CoV-2 variants of concern on cellular immune response should also be addressed in future research. It has been suggested that the resolution of SARS-CoV-2 infection and COVID-19 is significantly dependent on CD4+ and CD8+ T-cell responses [126], which also play a role in modulating disease severity [45,127]. In convalescent individuals, T-cell immunity is not restricted to spike-derived epitopes, and thus, it would be reasonable to assume that it would remain largely intact for new variants.
However, in recipients of the majority of currently available vaccines, which offer S protein as the target immunogen, T-cell immunity is limited to spike epitopes. Therefore, it is of essence to determine whether new variant mutations in these epitopes impair T-cell responses in a similar way as escape from neutralizing antibodies.
However, studies dealing with this problem are still scarce, mainly because measurements of T-cell immunity are more challenging for routine clinical practice than antibody detection assays. So far, the effect of variants B.1.1.7, B.1.351, P.1, and B.1.427/29 was found to be negligible on both CD4+ and CD8+ T-cell responses in the recipients of mRNA-based vaccines [128,129]. This was supported by the result of completely conserved epitopes for 93% of CD4+ and 97% of CD8+ T-cells in the variants.
In addition, it was pointed out that HLA binding capacity is not affected in the majority of cases by a single mutation in epitopes. However, the repertoire of recognized epitopes is probably substantially different from one individual to the other due to HLA polymorphism, and thus, the negative impact of the mutations of specific variants on each single person could not be entirely dismissed [128].
The incidence of the B.1.427/B.1.429 lineages is increasing rapidly
The SARS-CoV-2 B.1.427/B.1.429 variant was reported for the first time at the beginning of 2021 in California and as of May 2021 has been detected in 34 additional countries (41, 42). The two lineages B.1.427 and B.1.429 (belonging to clade 20C according to Nextstrain designation) share the same S mutations (S13I, and W152C in the NTD and L452R in the RBD), but harbor different mutations in other SARS-CoV-2 genes (42).
Molecular clock analysis suggest that the progenitor of both lineages emerged in May 2020, diverging to give rise to the B.1.427 and B.1.429 lineages in June-July 2020 (42). The fast rise in the number of cases associated with the B.1.427/B.1.429 lineages led to their classification as a VOC by the US Center for Disease Control (https://www.cdc.gov/coronavirus/2019-ncov/cases-updates/variant-surveillance/variant-info.html).
As of April 30, 2021, 8,441 and 21,072 sequenced genomes are reported in GISAID for the B.1.427 and B.1.429 lineages, respectively. This VOC was detected in California and in other US states, and more recently in 34 additional countries worldwide (Fig. 1, A to H, and table S1). The number of B.1.427/B.1.429 genome sequences deposited increased rapidly after December 2020, with an incidence exceeding 50% in California since February 2021 (Fig. 1, B to E).
Collectively, this analysis illustrates the increased incidence of the B.1.427/B.1.429 VOC, and its progressive geographical spread from California to other US states and other countries, which is consistent with a recent study suggesting enhanced transmissibility relative to the ancestral isolate (42).

(A) World map showing the geographic distribution and sequence counts of B.1.427/B.1.429 VOC as of April 30, 2021. (B) Cumulative and individual B.1.427/B.1.429 VOC sequence counts by month. (C to E) Total number of SARS-CoV-2 (grey) and B.1.427/B.1.429 VOC (blue/orange) sequences deposited on a monthly basis worldwide (C), in the US (D) and in California (E). (F to H) Total number of B.1.427/B.1.429 (F), B.1.427 (G) and B.1.429 (H) sequences deposited by country as of April 30, 2021. Only countries with n≥2 deposited sequences are shown.
B.1.427/ B.1.429 S reduces sensitivity to vaccine-elicited Abs
To assess the impact of the three mutations present in the B.1.427/B.1.429 S glycoprotein on neutralization, we first compared side-by-side the neutralization potency of mRNA vaccine-elicited Abs against G614 S and B.1.427/B.1.429 S pseudoviruses. We used plasma from fifteen individuals who received two doses of Moderna mRNA-1273 vaccine and from fifteen individuals who received two doses of Pfizer/BioNtech BNT162b2 vaccine collected between 7 and 27 days after booster immunization (table S2).
All vaccinees had substantial plasma neutralizing activity against G614 SARS-CoV-2 S pseudotyped viruses. Using a murine leukemia virus (MLV) pseudotyping system, geometric mean titers (GMTs) showed that the average neutralization potency of the Moderna mRNA1273-elicited plasma was reduced 2.4-fold for B.1.427/B.1.429 S (GMT: 178) compared to G614 S (GMT: 424) (Fig. 2, A and B; figs. S1 and S2; and table S3) whereas it was reduced 2.3-fold with Pfizer/BioNtech BNT162b2-elicited plasma (B.1.427/B.1.429 GMT: 78 versus G614 GMT: 182) (Fig. 2, C and D; figs. S1 and S2; and table S3).
Using a vesicular stomatitis virus (VSV) pseudotyping system, we observed a 2.2-fold average reduction of Moderna mRNA1273-elicited plasma neutralizing activity against B.1.427/B.1.429 S (GMT: 213) compared to G614 S (GMT: 464) pseudoviruses (Fig. 2, E and F; figs. S1 and S2; and table S3) and a 2.5-fold average reduction of Pfizer/BioNtech BNT162b2-elicited plasma neutralizing activity against B.1.427/B.1.429 S (GMT: 113) compared to G614 S (GMT: 285) pseudoviruses (Fig. 2, G and H; figs. S1 and S2; and table S3).
We also analyzed plasma from 18 individuals, 5 of whom were previously infected with wildtype SARS-CoV-2, who received two doses of Pfizer/BioNtech BNT162b2 vaccine and whose samples were collected between 14 and 28 days after booster immunization. We compared the neutralization potency of Pfizer/BioNtech BNT162b2 vaccine-elicited Abs against D614 S, B.1.427/B.1.429 S, B.1.1.7 S, B.1.351 S and P.1 S VSV pseudotyped viruses using Vero E6 expressing TMPRSS2 as target cells.
GMTs plasma neutralization potency was reduced 2.9-fold for B.1.427/B.1.429 S (GMT: 197) compared to D614 S (GMT: 570), which is a comparable decrease to that observed with B.1.351 (GMT: 180, 3.2-fold reduction) and greater to that observed with B.1.1.7 and P.1 (GMT: 450 and 330, 1.3-fold and 1.7-fold reduction, respectively) pseudotyped viruses (Fig. 2, I and J; figs. S1 and S2; and table S3). These data indicate that the three B.1.427/B.1.429 S residue substitutions lead to a modest but significant reduction of neutralization potency from vaccine-elicited Abs.

(A, B, E, and F) Neutralizing Ab titers (ID50) shown as pairwise connected [(A) and (E)] or the geometric mean titer, GMT [(B) and (F)] against MLV [(A) and (B)] or VSV [(E) and (F)] pseudotyped viruses harboring G614 SARS-CoV-2 S or B.1.427/B.1.429 (B.1.429) S determined using plasma from individuals who received two doses of Moderna mRNA-1273 vaccine (blue). (C, D, G, and H) Neutralizing Ab titers (ID50) shown as pairwise connected [(C) and (G)] or the geometric mean titer, GMT [(D) and (H)] against MLV [(C) and (D)] or VSV [(G) and (H)] pseudotyped viruses harboring G614 SARS-CoV-2 S or B.1.427/B.1.429 (B.1.429) S determined using plasma from individuals who received two doses of Pfizer/BioNtech BNT162b2 mRNA vaccine (red). (I and J) Neutralizing Ab ID50 (I) and GMT (J) titers against VSV pseudotyped viruses harboring D614 SARS-CoV-2 S, B.1.427/B.1.429 S, B.1.1.7 S, B.1.351 S, or P.1 S determined using plasma from naïve (blue) and previously infected (red) individuals who received two doses of Pfizer/BioNtech BNT162b2 mRNA vaccine. Naïve: vaccinated individuals who had not been previously infected with SARS-CoV-2. Immune: vaccinated individuals who had been previously infected with SARS-CoV-2. (K and L) Neutralizing Ab ID50 (K) and GMT (L) titers against VSV pseudotyped viruses harboring D614 SARS-CoV-2 S, B.1.427/B.1.429 S, B.1.1.7 S, B.1.351 S or P.1 S determined using plasma from convalescent individuals who were infected with wildtype SARS-CoV-2. Neutralization data shown in (A) to (H) and (I) to (L) were performed using 293T-ACE2 and VeroE6-TMPRSS2, respectively. Data are average of n = 2 replicates.
We also analyzed plasma from 9 convalescent donors, who experienced symptomatic COVID-19 in early 2020 (and consequently were likely exposed to the Wuhan-1 or a closely related SARS-CoV-2 isolate) collected 15 to 28 days post symptom onset (table S2). The neutralization potency of the 9 convalescent donor plasma was reduced 3.4-fold for B.1.427/B.1.429 S (GMT: 70) compared to G614 S (GMT: 240), similar to what we observed with B.1.351 (4.4-fold, GMT: 55) and P.1 (3.3-fold, GMT: 72) pseudotyped viruses, whereas neutralization of B.1.1.7 was less affected (1.9-fold, GMT: 127) (Fig. 2, K and L; figs. S1 and S2; and table S3). In several cases the level of neutralizing activity against the VOC was found to be below the limit of detection.
These findings show that the three mutations present in the B1.427/B.1.429 S glycoprotein decrease the neutralizing activity of vaccine-elicited and infection-elicited Abs, suggesting that these lineage-defining residue substitutions are associated with immune evasion. However, these data also underscore the higher quality of Ab responses induced by vaccination compared to infection and their enhanced resilience to mutations found in VOC.
B.1.427/B.1.429 S mutations reduce sensitivity to RBD- and NTD-specific Abs
To evaluate the contribution of RBD and NTD substitutions to the reduced neutralization potency of sera from vaccinees and convalescent plasma, we compared the neutralizing activity of 34 RBD and 10 NTD mAbs against the D614 S or B.1.427/B.1.429 S variant using a VSV pseudotyping system (1, 43).
The panel of RBD-specific mAbs (including 6 clinical mAbs) recognize distinct antigenic sites as previously characterized (10, 11, 13, 20, 44, 45). Briefly, epitopes span the RBM (antigenic sites Ia and Ib), a cryptic antigenic site II, the exposed N343 glycan-containing antigenic site IV and a second cryptic antigenic site V (10, 11).
A total of 14 out of 34 mAbs showed a reduced neutralization potency when comparing B.1.427/B.1.429 S and D614 S pseudoviruses (Fig. 3, A to C, and fig. S3). Considering the 6 mAbs in clinical use: regdanvimab (CT-P59), and to a smaller extent etesevimab (LY-CoV016), showed a reduction in neutralization potency, whereas bamlanivimab (LY-CoV555) entirely lost its neutralizing activity.
Neutralization mediated by the casirivimab/imdevimab mAb cocktail (REGN10933 and REGN10987) (14, 15), and by VIR-7831 (derivative of S309, recently renamed sotrovimab) (10, 23, 24), is unaffected by the B.1.427/B.1.429 S variant. To address the role of the L452R mutation in the neutralization escape from RBD-specific Abs, we tested the binding of the 34 RBD-specific mAbs to WT and L452R mutant RBD by biolayer interferometry (fig. S4). The 10 RBD-specific mAbs experiencing a 10-fold or greater reduction in neutralization potency of the B.1.427/B.1.429 variant, relative to D614 S, bound poorly to the L452R RBD mutant, demonstrating a direct role of this mutation im immune evasion.

(A and D) Neutralization of SARS-CoV-2 pseudotyped VSV carrying D614 (grey) or B.1.427/B.1.429 (orange) S protein by clinical-stage RBD mAbs (A) and an NTD-targeting mAb (S2X333) (D). Data are representative of n = 2 replicates. (B and E) Neutralization of SARS-CoV-2 S VSV pseudotypes carrying D614 or B.1.427/B.1.429 S by 34 mAbs targeting the RBD and 10 mAbs targeting the NTD. Data are the mean of 50% inhibitory concentration (IC50) values (ng/ml) of n = 2 independent experiments. Non-neutralizing IC50 titers were set at 105 ng/ml. (C and F) Neutralization by RBD-specific (C) and NTD-specific (G) mAbs showed as mean IC50 values (top) and mean fold change (bottom) for B.1.427/B.1.429 S (orange) relative to D614G S (grey) VSV pseudoviruses. VIR-7831 is a derivative of S309 mAb (sotrovimab). *, VIR-7832 (variant of VIR-7831 carrying the LS-GAALIE Fc mutations) shown as squares. Non-neutralizing IC50 titers and fold change were set to 105 ng/ml and 104, respectively.
We found that the neutralizing activity of all 10 NTD-specific mAbs tested was abolished as a result of the presence of the S13I and W152C mutations (Fig. 3, D to F). These data indicate that the decreased potency of neutralization of the B.1.427/B.1.429 variant results from evasion of both RBD- and NTD-specific mAb-mediated neutralization.
Structural characterization of the SARS-CoV-2 B.1.427/B.1.429 S trimer
To visualize the changes in SARS-CoV-2 B.1.427/B.1.429 S that contribute to immune evasion, we determined a cryoEM structure of the variant S ectodomain trimer (carrying the HexaPro mutations (46)) bound to the RBD-specific mAb S2M11 and the NTD-specific mAb S2L20 at 2.3 Å resolution (Fig. 4A, fig. S5, and table S4). S2M11 was used to lock the RBDs in the closed state (Fig. 4B) whereas S2L20 was used to stabilize the NTDs (Fig. 4C) (12, 13). Superimposing the regdanvimab- (CT-P59) and bamlanivimab- (LY-CoV555) bound SARS-CoV-2 RBD structures to B.1.427/B.1.429 S reveal that the introduced L452R is sterically incompatible with binding of these mAbs (Fig. 4, D and E), rationalizing the dampening or loss of neutralizing activity.

(A) Structure of the S trimer (surface rendering) bound to the S2M11 and S2L20 Fabs (ribbons) in two orthogonal orientations. SARS-CoV-2 S protomers are colored pink, cyan, and gold, whereas the S2L20 Fab heavy and light chains are colored dark and light green, respectively, and the S2M11 Fab heavy and light chains are colored dark and light gray, respectively. Only the Fab variable domains are resolved in the map. N-linked glycans are rendered as dark blue spheres. (B) Zoomed in view of the S2M11-bound RBD with R452 shown in ball and stick representation. (C) Zoomed in view of the S2L20-bound NTD with disordered N terminus, supersite β-hairpin and loop regions shown as dashed lines. (D) Superimposition of the CT-P59–bound SARS-CoV-2 RBD structure (PDB 7CM4) on the SARS-CoV-2 B.1.427/B.1.429 S cryoEM structure show that R452 would sterically clash with the mAb. (E) Superimposition of the LY-CoV555–bound SARS-CoV-2 RBD structure (PDB 7KMG) on the SARS-CoV-2 B.1.427/B.1.429 S cryoEM structure show that L452R would sterically clash with the mAb. (F) Superimposition of the S2X333-bound SARS-CoV-2 S structure (PDB 7LXW) on the SARS-CoV-2 B.1.427/B.1.429 S cryoEM structure reveals that most of the NTD antigenic supersite epitope residues are disordered. (G) Superimposition of the ACE2-bound SARS-CoV-2 RBD structure (PDB 7DMU) on the SARS-CoV-2 B.1.427/B.1.429 S cryoEM structure show that L452R points away from the interface with ACE2
We subsequently used local refinement to account for the conformational dynamics of the NTD and S2L20 relative to the rest of S and obtained a cryoEM reconstruction of the NTD bound to S2L20 at 3.0 Å resolution (Fig. 4C, fig. S5, and table S4). The structure reveals that the B.1.427/B.1.429 NTD antigenic supersite is severely altered. The N terminus is disordered up to residue 27, as is the supersite β-hairpin (disordered between residues 137-158) and the supersite loop (disordered between residues 243-264) (Fig. 4F). These structural changes explain the abrogation of binding and neutralization of the panel of NTD-specific mAbs evaluated.
Overlaying an ACE2-bound SARS-CoV-2 RBD structure with the B.1.427/B.1.429 variant S structure shows that the R452 residue points away from and does not contact ACE2, suggesting that this substitution would not affect receptor engagement (Fig. 4G). We next evaluated binding of the monomeric human ACE2 ectodomain to immobilized B.1.427/B.1.429 and wildtype RBDs using surface plasmon resonance (fig. S6, A and B, and table S5) biolayer interferometry (fig. S6, C to E, and table S5) as well as binding of B.1.427/B.1.429, B.1.1.7 and wildtype RBDs to immobilized human ACE2 by ELISA (fig. S6F and table SS5). Our results indicate that the B.1.427/B.1.429 and wildtype RBDs bound to ACE2 with comparable affinities (whereas the B.1.1.7 RBD had a markedly increased affinity for ACE2 (34)), validating the structural observations.
Disulfide bond rearrangement in the B.1.427/B.1.429 variant NTD antigenic supersite
To investigate further the molecular basis for the loss of NTD-directed mAb neutralizing activity and structural changes in the NTD, we analyzed binding of a panel of NTD-specific mAbs to recombinant SARS-CoV-2 NTD variants using ELISA. The S13I signal peptide mutation dampened binding of 5 mAbs and abrogated binding of 5 additional mAbs out of 11 neutralizing mAbs evaluated (Fig. 5A and fig. S7).
Furthermore, the W152C mutation reduced recognition of 6 NTD neutralizing mAbs, including a complete loss of binding for two of them, with a complementary pattern to that observed for S13I (Fig. 5A and fig. S7). The B.1.427/B.1.429 S13I/W152C NTD did not bind to any NTD-directed neutralizing mAbs, which are known to target a single antigenic site (antigenic site i) (12), whereas binding of the non-neutralizing S2L20 mAb to the NTD antigenic site iv was not affected by any mutants, confirming proper retention of folding, as supported by the structural data (Fig. 5A and fig. S7).
Binding of vaccine-elicited plasma to NTD mutants confirmed and extended these observations with polyclonal Abs by showing an increasingly marked reduction in binding titers due to the W152C, S13I and S13I/W152C residue substitutions (Fig. 5B and fig. S8).

(A) Binding of a panel of 11 neutralizing (antigenic site i) and 1 non-neutralizing (antigenic site iv) NTD-specific mAbs to recombinant SARS-CoV-2 NTD variants analyzed by ELISA displayed as a heat map. (B) Binding of plasma Abs from vaccinated individuals to recombinant SARS-CoV-2 NTD variants analyzed by ELISA. The mean dilution factor for each mutant was compared by the one-way ANOVA test against wildtype yielding p values < 0.05 () and <0.001 (*). (C to G) Deconvoluted mass spectra of purified NTD constructs, including the wildtype NTD with the native signal peptide (B), the S13I NTD (C), the S13I and W152C NTD (D), the W152C NTD (E), and the S12F NTD (F). The empirical mass (black) and theoretical mass (red) are shown beside the corresponding peak. Additional 119 Da were observed for the S13I and W152C NTDs corresponding to cysteinylation of the free cysteine residue in these constructs (as L-cysteine was present in the expression media). The cleaved signal peptide (blue text) and subsequent residue sequence (black text) are also shown based on the MS results. Mutated residues are shown in bold. Cysteines are highlighted in light orange (unless in the cleaved signal peptide) while disulfide bonds are shown as dotted light orange lines between cysteines. Residues are numbered for reference.
We previously showed that disruption of the C15/C136 disulfide bond that connects the N terminus to the rest of the NTD, through mutation of either residue or alteration of the signal peptide cleavage site, abrogates the neutralizing activity of mAbs targeting the NTD antigenic supersite (site i) (12).
As the S13I substitution resides in the signal peptide and is predicted to shift the signal peptide cleavage site from S13-Q14 to C15-V16, we hypothesized that this substitution indirectly affects the integrity of NTD antigenic site i, which comprises the N terminus. Mass spectrometry analysis of the S13I and S13I/W152C NTD variants confirmed that signal peptide cleavage occurs immediately after residue C15 (Fig. 5, C to E).
As a result, C136, which would otherwise be disulfide linked to C15, is cysteinylated in the S13I NTD due to the presence of free cysteine in the expression media (Fig. 5D and fig. S9). Likewise, the W152C mutation, which introduces a free cysteine, was also found to be cysteinylated in the W152C NTD (Fig. 5E). It is not clear if cysteinylation would occur during natural infection with S13I or W152C mutants alone, or what contribution cysteinylation plays in immune evasion of S13I or W152C mutants alone. Notably, dampening of NTD-specific neutralizing mAb binding is stronger for the S13I mutant than for the S12P mutant which we previously showed to also shifts the signal peptide cleavage site to C15-V16 (Fig. 5A).
Conversely, we did not observe any effect on mAb binding of the S12F substitution, which has also been detected in clinical isolates, in agreement with the fact that this mutation did not affect the native signal peptide cleavage site (i.e., it occurs at the S13-Q14 position), as observed by mass spectrometry (Fig. 5G). In the absence of the C15-C136 disulfide bond the N terminus is no longer stapled to the NTD, consistent with the structural data showing that the N terminus of the B.1.427/B.1.429 variant becomes disordered relative to the rest of the NTD (Fig. 4C).
Although the S13I and W152C NTD variants were respectively cysteinylated at positions C136 and W152C, the double mutant S13I/W152C was not cysteinylated, suggesting that C136 and W152C had formed a new disulfide bond. (Fig. 5, D to F). Tandem mass-spectrometry analysis of non-reduced, digested peptides identified linked discontinuous peptides containing C136 and W152C (fig. S9) confirming that a disulfide bond forms between C136 and W152C in the S13I/W152C NTD of the B.1.427/B.1.429 variant. W152C is in the β-hairpin of the antigenic supersite, and the formation of a new disulfide bond with C136 would move residues in the β-hairpin >20 Å and the local structure of the β-hairpin was disordered in the B.1.427/B.1.429 variant (Fig. 4C).
Collectively, these findings demonstrate that the S13I and W152C mutations found in the B.1.427/B.1.429 S variant are jointly responsible for escape from NTD-specific mAbs, due to deletion of the SARS-CoV-2 S two N-terminal residues and overall rearrangement of the NTD antigenic supersite. Our data support that the SARS-CoV-2 NTD evolved a compensatory mechanism to form an alternative disulfide bond and that mutations of the S signal peptide occur in vivo in a clinical setting to promote immune evasion. The SARS-CoV-2 B.1.427/B.1.429 S variant therefore relies on an indirect and unusual neutralization-escape strategy.
reference link : https://science.sciencemag.org/content/early/2021/06/30/science.abi7994
reference link: https://www.mdpi.com/1999-4915/13/7/1192/htm



{kind=link}