









 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Nonostante il lento tasso di evoluzione del SARS-CoV-2 rispetto ad altri virus a RNA, la sua trasmissione massiccia e rapida durante la pandemia di COVID-19 gli ha permesso di acquisire una significativa diversità genetica da quando è entrato per la prima volta nella popolazione umana.
Ciò ha portato all’emergere di numerose varianti, alcune delle quali recentemente etichettate come “varianti preoccupanti” (VOC), a causa del loro potenziale impatto sulla trasmissione, morbilità/mortalità e l’elusione della neutralizzazione da parte degli anticorpi provocati da infezione, vaccinazione o applicazione terapeutica.
Il potenziale per eludere la neutralizzazione è il risultato della diversità degli epitopi bersaglio generati dall’accumulo di mutazioni nella proteina spike. Mentre tre COV riconosciuti a livello mondiale (Alpha o B.1.1.7, Beta o B.1.351 e Gamma o P.1) rimangono sensibili alla neutralizzazione sebbene a livelli ridotti da parte dei sieri di individui convalescenti e destinatari di diversi vaccini anti-COVID19, l’effetto della variabilità degli spike è molto più evidente sulla capacità di neutralizzazione degli anticorpi monoclonali.
Il nuovo VOC Delta o lignaggio B.1.617.2, così come i VOC accettati localmente (Epsilon o B.1.427/29-US e B1.1.7 con E484K-UK) indicano la necessità di un attento monitoraggio delle nuove varianti su un livello globale. Le caratteristiche dei VOC, i loro modelli mutazionali e il ruolo che le mutazioni giocano nell’evasione immunitaria sono riassunti in questa recensione.
Organizzazione del genoma SARS-CoV-2 e della proteina Spike
SARS-CoV-2 appartiene all’ordine Nidovirales, famiglia Coronaviridae, sottofamiglia Orthocoronavirinae e genere Betacoronavirus. I virioni dei coronavirus sono sferici con diametri medi da 80 a 120 nm. Sono avvolti da genomi di RNA a singolo filamento (ss) positivi. L’analisi genomica di tre coronavirus appena scoperti ha mostrato che SARS-CoV-2 ha una somiglianza di sequenza del 79% e del 50% rispettivamente con SARS-CoV e MERS-CoV [8,9].
Il coronavirus con il genoma più simile a SARS-CoV-2 è il virus del pipistrello a ferro di cavallo RaTG13 Rhinolophus affinis con il 96% di somiglianza [8,10] Il
genoma di SARS-CoV-2 è sotto forma di ssRNA con senso positivo e una lunghezza di circa 30.000 nucleotidi. Questo genoma non segmentato include una regione 5′-non tradotta (UTR), seguita dal complesso di replicasi (ORF1a e ORF1ab), geni strutturali per spike (S), busta (E), membrana (M), proteine nucleocapside (N), e diversi open reading frame (ORF) per proteine accessorie inserite tra quattro geni strutturali, che terminano con 3′-UTR con coda di poli A [1,11] (Figura 1).
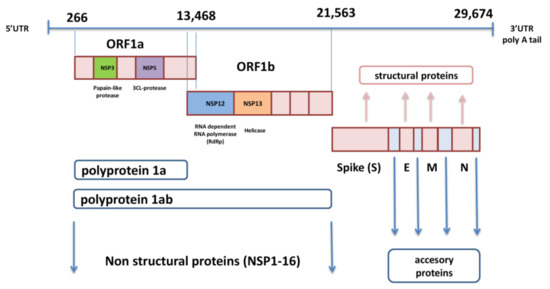
I geni ORF1a e ORF1b, situati uno accanto all’altro vicino al 5′-UTR, occupano i due terzi del genoma di SARS-CoV-2 e codificano per le poliproteine pp1a e pp1ab. Queste due poliproteine sono scisse con l’enzima autoproteolitico in 16 proteine non strutturali (nsp1-16) che sono coinvolte nella replicazione virale, nella trascrizione, nell’immunomodulazione, nella transattivazione genica e nella resistenza alla risposta antivirale innata [12]. L’ultimo terzo del genoma contiene geni per proteine strutturali e accessorie.
Il gene S codifica la glicoproteina spike, che è la proteina più importante del virione e consente l’ingresso virale nella cellula bersaglio [13]. La glicoproteina M contiene tre domini, dominio C terminale, transmembrana e N terminale, ed è necessaria per l’assemblaggio e il germogliamento dei virioni [14].
La proteina dell’involucro include anche tre domini e svolge un ruolo importante nella patogenesi dell’infezione da COVID-19 perché il suo dominio C-terminale si lega alla proteina a giunzione stretta umana PALS1 [15]. Il nucleocapside si lega all’RNA virale e influenza le prestazioni di replicazione di SARS-CoV-2 [16]. Le proteine accessorie contribuiscono in modo significativo all’evasione della risposta immunitaria innata interferendo con la sintesi dell’interferone (IFN) e ostruendo le vie del segnale all’interno della cellula [17].
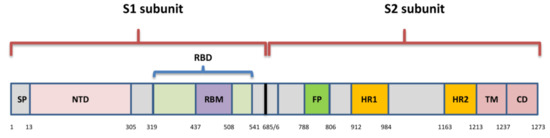
Il picco è una glicoproteina transmembrana lunga 1273 aminoacidi e a forma di omotrimero. Comprende il dominio di legame del recettore (RBD) che interagisce con l’enzima di conversione dell’angiotensina 2 (ACE2) del recettore della cellula ospite [18]. La proteina SARS-Cov-2 S condivide la somiglianza della sequenza di amminoacidi del 76,7-77% con SARS-CoV di esseri umani e zibetti, 75-98% con coronavirus di pipistrello dello stesso sottogenere (Sarbecovirus) e 90,7-92,6% con coronavirus di pangolino [8].
Spike include due subunità: S1 (aa 1–685) e S2 (aa 686–1273) (Figura 2). La subunità S1 comprende un dominio N-terminale (NTD) e RBD (aa 319–541), mentre la subunità S2 è composta da un peptide di fusione (FP) (chiamato anche subunità S2′), dominio eptapeptide 1 e 2 (HPD1, HPD2), dominio transmembrana (TM) e dominio citoplasmatico (CD) [19]. L’RBD è l’attore chiave all’interno della subunità S1 per l’attaccamento di SARS-CoV-2 ad ACE2 [20] ed è quindi un bersaglio molto importante per farmaci antivirali e anticorpi [21].
Contiene una struttura centrale e un motivo di legame al recettore (RBM) (aa 437-508), che è la parte più variabile della proteina spike che è importante per il legame alla superficie esterna di ACE2 [18]. C’è solo il 73% di somiglianza tra RBD di SARS-CoV-2 e SARS-CoV, sebbene entrambi si leghino all’ACE2 umano [22].
La caratterizzazione della stabilità meccanica del RBD di SARS-CoV-2 ha dimostrato che è più rigido rispetto a SARS-CoV, il che ha importanti conseguenze nel legame con ACE2, in quanto può resistere alle forze Browniane e cellulari pur rimanendo a stretto contatto durante le fasi iniziali dell’ingresso nella cella [23]. La proteina S persiste nella forma aperta e chiusa [24].
Nella forma chiusa, i motivi di riconoscimento sono nascosti, e nella forma aperta, il cosiddetto stato di accesso al recettore, RBD con RBM sono in conformazione verso l’alto. Mentre sono nella conformazione up, sporgono dagli omotrimeri e consentono il legame al recettore, il processo di fusione e l’ingresso del virus nella cellula ospite [20]. La fusione di due membrane è il passaggio cruciale nel ciclo di vita virale [25].
È stato suggerito che alcuni residui chiave sono cruciali per la stabilizzazione della proteina spike durante le transizioni da conformazioni chiuse a conformazioni aperte prima del riconoscimento di ACE2 [26]. Tra l’NTD e l’RBD si formano diversi contatti ad alta frequenza che sono responsabili della stabilità conformazionale locale e svolgono un ruolo durante la transizione dallo stato chiuso a quello aperto.
La subunità S2 ha tre stati conformazionali: (1) stato nativo di prefusione; (2) stato intermedio pre-tornante; e (3) stato a forcina post-fusione. Le subunità S1 e S2, che sono legate in modo non covalente nello stato di prefusione, hanno un ruolo unito nel legarsi al recettore della cellula ospite [22].
La subunità S1 interagisce con il recettore e consente l’attaccamento del virione [27], mentre la subunità S2 è coinvolta nel processo di fusione. La proteina S viene scissa dalle proteasi dell’ospite, dalle serina proteasi transmembrana di tipo II (TMPRSS2) e dalla furina, alla giunzione di S1 e S2 in un sito di scissione polibasico (S2′) [28,29]. La scissione nel sito S2′ attiva le proteine, che inducono cambiamenti conformazionali irreversibili nella proteina S, cruciale per la fusione delle membrane virali e cellulari [24].
L’inserimento di quattro amminoacidi (PRRA) nel sito di scissione polibasico rappresenta una caratteristica genomica specifica di SARS-CoV-2 [28]. Questo sito non è osservato nei coronavirus correlati tranne che in un coronavirus derivato da pipistrello da Rhinolophus malayanus (RmYN02), che ha un’inserzione di tre amminoacidi (PAA) [30]. Alcuni studi indicano che questo sito di scissione della furina induce l’instabilità di SARS-CoV-2, causando cambiamenti conformazionali necessari per il legame di RBD al recettore [31].
Risposta immunitaria a SARS-CoV-2
I virus, come SARS-CoV-2, oltre all’immunità innata, inducono una risposta immunitaria adattativa sia umorale che cellulare, innescando diversi meccanismi di difesa per combattere l’infezione acuta. Le cellule NK, i monociti (macrofagi) e l’IFN di tipo I sono cruciali nella risposta a questo virus. Una diminuzione del numero di cellule NK [32] e cellule dendritiche plasmacitoidi (fonte principale di IFN tipo I) e monociti che producono interleuchina (IL)-6 dominante sono caratteristici di una risposta immunitaria innata SARS-CoV-2 inappropriata [33]. https://pubmed.ncbi.nlm.nih.gov/33357409/Large (19 giugno 2021), come RECOVERY, hanno stabilito che l’anti-IL-6 in combinazione con steroidi è una potenziale opzione per i pazienti ipossici con evidenza di iperinfiammazione [34].
Ci sono dati che mostrano che esiste una correlazione statisticamente significativa per alcune varianti comuni in tre geni legati all’immunità innata, MBL2, TMPRSS2 e CD27. MBL2 codifica per una proteina C legante il mannosio che si lega al mannosio, attivando la via del complemento della lectina; TMPRSS2 scinde la proteina spike e garantisce l’internalizzazione virale [35], mentre il recettore CD27 è necessario per la generazione e il mantenimento a lungo termine dell’immunità delle cellule T.
Ci sono alcuni dati che suggeriscono che l’immunità innata addestrata potrebbe anche avere un ruolo nella protezione contro COVID-19 [36,37]. Diversi studi clinici stanno studiando se vaccini non correlati, come il vaccino contro il morbillo, la parotite, la rosolia e il vaccino BCG, possono provocare un’immunità innata addestrata e migliorare la protezione contro COVID-19 [38].
È importante sottolineare che il recupero dall’infezione da COVID-19 è legato a un’adeguata risposta immunitaria e che la gravità della malattia è correlata alla reazione immunitaria compromessa. È noto che questo virus con il suo particolare potenziale di inattivazione della risposta basata sull’IFN porta all’indebolimento dell’immunità innata.
Inoltre, una volta presente nella cellula ospite, il SARS-CoV-2 attiva la famiglia dei recettori NOD-like, inducendo la formazione di un inflammasoma. Ciò contribuisce al rilascio delle citochine pro-infiammatorie, IL-1, IL-6, TNF e IL-18. La via NF-kB viene attivata dopo l’interazione dell’RNA virale con i recettori Toll-like e migliora la produzione di citochine pro-infiammatorie. Pertanto, l’infiammazione inizia e porta al rilascio di un numero di citochine dalle cellule immunitarie attivate e si verifica la cosiddetta tempesta di citochine, che può essere pericolosa per la vita [39].
La risposta immunitaria umorale a SARS-CoV-2 è mediata da anticorpi specifici principalmente per la glicoproteina spike, tutte le sue parti compreso NTD e la proteina nucleocapside [40]. Questi anticorpi neutralizzano il legame virale alle cellule che esprimono i recettori ACE2 e l’infezione di queste cellule [41]. Molti studi che esaminano la durata della protezione mediante anticorpi neutralizzanti funzionali e il potenziale di reinfezione hanno dimostrato che la maggior parte dei pazienti con COVID-19 ha risposte IgM, IgA e IgG virus-specifiche nei giorni successivi all’infezione [42].
Negli individui con COVID-19 lieve, in diversi studi è stato osservato un rapido declino dei titoli IgG specifici per RBD entro 2-4 mesi, suggerendo che l’immunità umorale indotta da SARS-CoV-2 potrebbe non essere di lunga durata in individui con lieve malattia [43]. I titoli anticorpali erano significativamente più alti nei pazienti con malattia grave rispetto ai pazienti con malattia lieve ed erano associati a esiti clinici [44].
Tuttavia, uno studio completo sull’immunità adattativa alla SARS-CoV-2, che ha anche esaminato l’associazione con la gravità della malattia, ha mostrato che la concentrazione di anticorpi neutralizzanti non era correlata alla gravità del COVID-19 [45]. Non esiste un’immunità preesistente alla SARS-CoV-2 nella popolazione, se non attraverso la reattività crociata con altri coronavirus [46]. È anche importante valutare le cellule B della memoria oltre alla misurazione degli anticorpi per caratterizzare meglio l’immunità umorale. Sebbene alti titoli circolanti di anticorpi neutralizzanti siano comuni surrogati dell’immunità protettiva, ci sono molte situazioni in cui gli anticorpi circolanti non raggiungono livelli sufficienti ed è necessario un input aggiuntivo dalle cellule B della memoria [47].
Se gli anticorpi circolanti scompaiono nel tempo, i dati suggeriscono che è probabile che le cellule B della memoria robuste forniscano una rapida fonte di anticorpi protettivi in caso di potenziale reinfezione da SARS-CoV-2. Inoltre, nell’infezione con varianti che possono parzialmente sfuggire alla neutralizzazione da parte degli anticorpi circolanti presenti [48,49,50], saranno necessarie cellule B della memoria vitale per rientrare nei centri germinali e trasformarsi per rispondere a nuovi epitopi spike [51 ].
Inoltre, sono stati isolati anticorpi monoclonali umani (mAb) mirati sia a NTD che a RBD di SARS-CoV-2, con quelli che hanno come bersaglio RBD particolarmente potenti. Questi anticorpi sono utilizzati clinicamente [52,53], in modalità terapeutiche e profilattiche. Inoltre, la selezione di miscele di anticorpi con mutazioni di fuga non sovrapposte dovrebbe aiutare e prolungare l’efficacia delle terapie anticorpali nell’infezione da SARS-CoV-2 [54].
Dopo l’infezione, un certo numero di cellule T HLA-DR+, sia CD4+ che CD8+, aumenta nei primi 7-10 giorni dopo i primi sintomi e diminuisce dopo tre settimane [55,56,57]. La risposta delle cellule T CD4+ alla SARS-CoV-2 consiste principalmente di cellule T-helper-1 (Th1), che sono caratterizzate da un’elevata secrezione di IFN-γ e specificità per la glicoproteina spike strutturale, la proteina di membrana e la proteina nucleocapside . La risposta delle cellule T CD8+ specifica per SARS-CoV-2 ha prodotto anche IFN-γ e fattore di necrosi tumorale (TNF).
Le cellule T specifiche per SARS-CoV-2 esprimono perforina e granzimi dopo la riattivazione in vitro con antigeni virali. È stato anche dimostrato che durante la fase di convalescenza, le cellule T avevano un fenotipo di memoria, sia le cellule T CD4+ che CD8+ che esprimevano IFN-γ, IL-2 e TNF [58]. La risposta delle cellule T follicular helper (Tfh) è cruciale per lo sviluppo di una forte immunità umorale attraverso la formazione di centri germinali e la fornitura di co-stimolazione (interazione CD40-CD40-L e citochine) alle cellule B [59].
Uno studio sul sequenziamento dell’RNA a singola cellula delle cellule T CD4+ specifiche per SARS-CoV-2 ha rilevato un aumento della percentuale di cellule Tfh nei pazienti con malattia grave [60]. Altri fattori di rischio per COVID-19 grave sono l’aumento del numero di cellule Th17, le cellule T che esprimono marcatori di esaurimento (come PD-1) e l’esaurimento delle cellule T sia αβ che [61]. È anche possibile il riconoscimento degli antigeni SARS-CoV-2 da parte di cellule T preesistenti e cross-reattive create durante una precedente infezione con coronavirus umani [62].
Nel recente studio [63], gli autori hanno suggerito che la risposta delle cellule T e il legame degli anticorpi alla proteina spike inducono una protezione precoce nel COVID-19. Dopo i vaccini per l’mRNA, tutti gli individui sviluppano cellule T specifiche per gli spike, mentre l’80% sviluppa anticorpi che legano gli spike 10 giorni dopo la prima dose. Stanno suggerendo che la mancanza di anticorpi neutralizzanti non è essenziale per prevenire il COVID-19. Ad eccezione dei vaccini a virus interi uccisi, tutti i vaccini attuali offrono la proteina S come immunogeno bersaglio, limitando l’immunità delle cellule T agli epitopi spike. Per gli epitopi delle cellule T, un’analisi dell’esposizione della popolazione ha proposto una serie di epitopi che si stima forniscano un’ampia copertura in tutto il mondo [64].
Variabilità genetica di SARS-CoV-2 e classificazione delle varianti
La diversità genetica di SARS-CoV-2 è il risultato di errori generati dalla sua RNA polimerasi RNA-dipendente (RdRp) e dalla ricombinazione [65]. La capacità dei coronavirus di ricombinarsi è associata alla capacità di commutazione del filamento di RdRp ed è probabile che abbia svolto un ruolo significativo nella loro evoluzione. Sebbene i coronavirus abbiano un tasso di mutazione più lento rispetto ad altri virus a RNA a causa della loro correzione di bozze di esoribonucleasi (nsp14), le conseguenze dell’accumulo di mutazioni sono ancora una delle principali preoccupazioni. È diventato ovvio che l’accumulo di mutazioni di aminoacidi potrebbe influenzare la trasmissibilità del virus, il suo tropismo cellulare e la sua patogenicità, presentando una seria sfida per l’efficienza dei vaccini e dei test diagnostici attuali.
Fino a poco tempo, la diversità osservata tra le sequenze SARS-CoV-2 era bassa. La prima mutazione della proteina spike D614G di SARS-CoV-2 in Europa è stata identificata nel gennaio 2020 in Germania [66]. Da allora, il ceppo che ospita D614G è diventato la variante pandemica dominante nella maggior parte dei paesi, probabilmente perché la mutazione ha consentito un relativo vantaggio di idoneità al ceppo originale di Wuhan e una maggiore infettività.
Il numero crescente di varianti di SARS-CoV-2 ha sviluppato la necessità della loro classificazione in gruppi come lignaggi e cladi. Il 31 maggio 2021, l’Organizzazione mondiale della sanità (OMS) ha introdotto nomi basati sull’alfabeto greco per varianti importanti al fine di semplificare la comunicazione pubblica intorno alle varianti e consentire il riferimento alle varianti in modo geograficamente neutro [67]. Tuttavia, questo non sostituisce i tre attuali sistemi di nomenclatura: GISAID (Global Initiative on Sharing All Influenza Data), Nextstrain e PANGO.
Esistono 8 cladi di SARS-CoV-2 o hCoV-19 (S, O, L, V, G, GH, GR e GV) identificati dal database GISAID [68], 11 cladi principali (19A, 19B e 20A-20I) riconosciuto da Nextstrain, mentre Rambaut et al. [69] e il team del software della Phylogenetic Assignment of Named Global Outbreak Lineage (PANGOLIN) hanno proposto 6 lignaggi principali (A, B, B.1, B.1.1, B.1.177, B.1.1.7) ora noti come PANGO nomenclatura.
Se una nuova variante emergente possiede marcatori genetici specifici che sono stati associati a maggiore trasmissibilità, morbilità e mortalità e capacità di eludere l’immunità naturale, nonché ridotta neutralizzazione da parte di anticorpi terapeutici o vaccinazione, ridotta efficacia dei trattamenti o potenziale impatto diagnostico, può essere etichettato come “variante in esame (VUI)” o “variante di interesse (VOI)” e se la sua prevalenza ed espansione supera il livello nazionale, può essere contrassegnato come “variante di interesse (VOC)”. Se c’è evidenza che una variante ha sviluppato caratteristiche che riducono significativamente l’efficacia delle misure di prevenzione o intervento esistenti, può essere definita una “variante con conseguenze elevate” [70,71,72].
Finora, ci sono quattro varianti di interesse riconosciute a livello globale: Alpha o lineage B.1.1.7 (Regno Unito), Beta o lineage B.1.351 (Sud Africa), Gamma o lineage P.1 (Giappone/Brasile) e Delta o stirpe B.1.617.2 (India) [70,71,72,73]. Un altro è stato riconosciuto come VOC nel Regno Unito e dall’ECDC – B.1.1.7 con E484K e altri due dagli Stati Uniti – Epsilon o B.1.427/29 [71,72,73].
L’alfa o il lignaggio B.1.1.7 (noto anche come 20I/501Y.V1 e VOC-202012/01) è emerso nel settembre 2020 nell’Inghilterra sudorientale. Ospita sette mutazioni missenso e tre residui cancellati nella proteina spike [74]. Grazie alla sua maggiore trasmissibilità, si è rapidamente diffuso in tutto il mondo ed è segnalato, dal 1 giugno 2021, in 160 paesi. Nel febbraio 2021, Public Health England (PHE) ha riconosciuto B.1.1.7 con mutazione E484K come nuovo VOC (VOC-202102/02) e da allora è stato identificato negli Stati Uniti. Tuttavia, questa variante non è stata rilevata nel Regno Unito dal marzo 2021, ma continua a diffondersi al di fuori del Regno Unito in base ai dati di sequenza.
Il beta o il lignaggio B.1.351 (noto anche come variante 20H/501Y.V2) è stato rilevato per la prima volta nella provincia del Capo orientale in Sudafrica alla fine del 2020 [75]. Contiene sette mutazioni e tre residui cancellati nella proteina spike. Questa variante è della massima preoccupazione per quanto riguarda la fuga immunitaria per le sue tre mutazioni all’interno del RBD e da allora è stata diffusa in 113 paesi. La variante Gamma o P.1 (nota anche come variante 20J/501Y.V3) derivante dal lignaggio B.1.1.28 è stata descritta per la prima volta in Brasile e Giappone nel dicembre 2020 e successivamente classificata come VOC a causa di 11 mutazioni spike, comprese le stesse tre in RBD come variante sudafricana [76].
Da allora è stato segnalato in 64 paesi. I lignaggi B.1.429, definiti da quattro e B.1.427 da due mutazioni spike, sono riconosciuti come VOC negli Stati Uniti e come VOI Epsilon dall’OMS [77]. Sono stati identificati per la prima volta in California (entrambi noti anche come CAL.20C e 20C/S:452R), dove hanno raggiunto una prevalenza di oltre il 50% a febbraio 2021. A giugno 2021, più di 60 paesi hanno segnalato casi causati da un variante appena riconosciuta: il lignaggio B.1.617 (noto anche come G/452R.V3) e i suoi tre sottolignaggi, i primi due rilevati nel dicembre 2020 e il terzo rilevato nel febbraio 2021 in India [70].
Tuttavia, da allora è diventato evidente che solo il sottolignaggio B.1.617.2 è associato a un maggiore rischio per la salute pubblica, motivo per cui ora è l’unico sottolignaggio di B.1.617 riconosciuto come VOC-Delta [67]. Il sottolignaggio B.1.617.1 è stato riclassificato in un VOI (variante Kappa) e, sebbene stia ancora dimostrando una maggiore trasmissibilità, la prevalenza globale sembra essere in declino. Sulla base dei rapporti, la prevalenza di B.1.617.3 è bassa e non è più classificata come VOC o VOI.
La principale speculazione sull’origine di nuove varianti con mutazioni accumulate propone che si siano evolute all’interno di pazienti immunosoppressi con infezione cronica che hanno sostenuto un’elevata replicazione virale per mesi e potrebbero essere stati trattati con plasma immunitario o anticorpi monoclonali [78,79,80]. Tuttavia, poiché i lignaggi di solito contengono mutanti intermedi circolanti, la diversità all’interno di alcuni lignaggi non può essere spiegata solo da una singola infezione a lungo termine in un individuo [75].
Implicazioni delle varianti SARS-CoV-2 nell’evasione immunitaria
Sebbene diversi lignaggi siano definiti da mutazioni in più di una regione del genoma, la massima attenzione è rivolta ai cambiamenti non sinonimi nel gene S, che possono alterare la proteina spike e influenzare il suo ruolo nell’ingresso del virus. Questo ruolo del picco lo ha determinato come bersaglio ideale per la risposta immunitaria e lo ha anche reso il bersaglio primario per la maggior parte dei vaccini attualmente approvati.
Sono stati osservati cambiamenti di aminoacidi attraverso l’intera proteina spike, ma la posizione esatta definisce l’impatto di ogni sostituzione. L’NTD e l’RBD sono le regioni più diverse e la maggior parte dei mAbs contro SARS-CoV-2 che sono stati caratterizzati colpiscono l’RBM, e alcuni sono specifici anche per il core e l’NTD dell’RBD [81]. I cambiamenti nei residui di spike all’interno dei principali epitopi possono ridurre o eliminare il legame e la neutralizzazione degli anticorpi, il che porterebbe a una ridotta efficacia degli anticorpi, derivati da infezioni naturali o vaccinazioni.
Tuttavia, si riscontrano cambiamenti anche all’interno del dominio C-terminale conservato della subunità S1 e S2. Queste regioni sono importanti per i cambiamenti conformazionali all’interno di S, che è necessario per l’attaccamento e la fusione virale e possono suscitare risposte neutralizzanti ancora sconosciute [82].
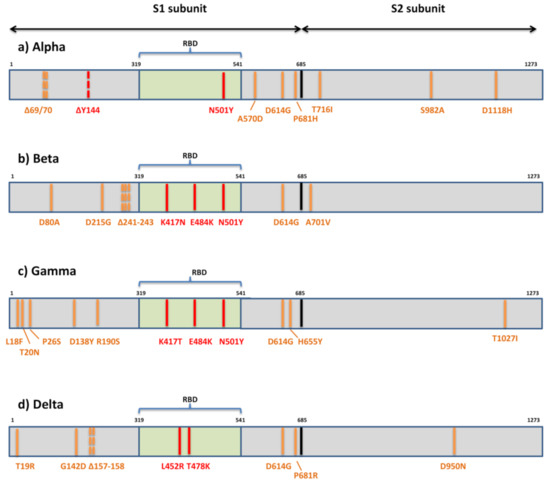
La prima variante Alpha o B.1.1.7 che ha sollevato preoccupazioni globali sull’aumento della trasmissibilità e sulla potenziale evasione immunitaria ospita sette mutazioni missenso (N501Y, A570D, D614G, P681H, T716I, S982A, D1118H) e tre delezioni in spike (69/70del e 144del) (Figura 3).
I tre residui eliminati si trovano all’interno di NTD, solo una mutazione (N501Y) è all’interno di RBM, tre sono visualizzati nel dominio C-terminale (CTD) di S1 e tre sono visualizzati all’interno di S2. Vari studi hanno finora dimostrato la ridotta potenza degli anticorpi neutralizzanti contro B.1.1.7 [49,83,84,85,86,87,88]. Questi studi condividono la conclusione generale che la variante B.1.1.7 rimane sensibile alla neutralizzazione, sebbene a livelli moderatamente ridotti, da parte di sieri di individui convalescenti e destinatari di diversi vaccini anti-COVID19.
La riduzione dei livelli di neutralizzazione è stata in media di 3 volte (da 1,5-10 volte) per i sieri convalescenti e ≈2 volte per i sieri dei soggetti vaccinati (basati su mRNA, vettori e subunità) 84,85,86]. Tuttavia, quando sono stati testati vari mAb contro questa variante, è stato dimostrato in modo uniforme che la variante B.1.1.7 può sfuggire alla neutralizzazione mediata da una frazione di anticorpi specifici per RBM e dalla maggior parte degli anticorpi specifici per NTD [49,83,88].
La spiegazione proposta per l’effetto più grave delle mutazioni spike sulla neutralizzazione da mAbs rispetto ai sieri è la policlonalità della neutralizzazione del siero [84]. È supportato dall’osservazione che una singola mutazione può diminuire il legame di un singolo mAbs ma non di altri anticorpi nello stesso cluster di legame. Una singola mutazione non può influenzare tutti gli anticorpi nello stesso cluster, poiché sembra che ogni anticorpo abbia un contatto molecolare ben definito e unico con lo stesso epitopo specifico. Pertanto, i sieri policlonali sono meno suscettibili ai cambiamenti nella neutralizzazione dovuti a una singola mutazione. I sieri policlonali contengono anche anticorpi non neutralizzanti il cui ruolo è ancora da chiarire.
La parte dell’effetto neutralizzante diminuito degli anticorpi contro la variante B.1.1.7 può essere attribuita all’unica mutazione RBM—N501Y. Si pensa che questa mutazione, condivisa da tre VOC riconosciuti a livello globale, sia il risultato dell’evoluzione adattativa virale [89] e ha dimostrato di aumentare l’affinità per ACE2 [85,90,91,92]. L’affinità di legame potenziata può contribuire a ulteriori interazioni con ACE2 consentite dal cambiamento 501 – i nuovi legami idrogeno ai residui 41 e 353 e anche a una conformazione più aperta dell’RBD [93,94].
Mentre alcuni riportano che il suo impatto antigenico è limitato a pochi mAb senza alcun effetto significativo sulla neutralizzazione da parte di sieri convalescenti o vaccinati [83], altri mostrano che l’aumento della trasmissione è combinato con la riduzione della potenza di neutralizzazione dei sieri convalescenti [85] . La spiegazione di ciò non risiede nel legame interrotto degli anticorpi all’RBM modificato, ma nella competizione degli anticorpi con ACE2 per il legame all’RBM. Pertanto, tutti i cambiamenti nell’RBM che conferiscono una maggiore affinità per ACE2 renderanno il virus più difficile da neutralizzare.
La significativa resistenza di B.1.1.7 alla neutralizzazione da parte di anticorpi specifici per NTD dovrebbe essere spiegata dalla presenza di tre residui deleti in questa regione. L’effetto di neutralizzazione di questi anticorpi può essere attribuito al ruolo che NTD ha nell’ingresso virale. Sebbene non sia stato ancora determinato per SARS-CoV-2, il NTD ha un ruolo nell’attaccamento alle cellule ospiti in diversi membri della famiglia CoV [95].
Per SARS-CoV-2, è stato proposto che NTD interagisca con i recettori ausiliari in tipi cellulari che non esprimono ACE2 (ad es. DC-SIGN/L-SIGN) [96]. La delezione NTD H69/V70 è osservata in B.1.1.7. e B.1.298 (visone danese) ma non è stato finora associato alla fuga da anticorpi specifici per NTD [88].
È stato dimostrato che una combinazione di del H69/V70 e N501Y aumenta l’infettività in vitro [97]. D’altra parte, è stato scoperto che la delezione Y144 annulla il legame con gli anticorpi neutralizzanti [49,52,88,98]. Non è ancora possibile determinare se le mutazioni NDT siano il risultato della selezione immunitaria o siano generate come parte del miglioramento dell’idoneità virale.
Altre mutazioni spike di B.1.1.7 appartengono al dominio C-terminale di S1 e S2 e finora non sono state percepite per influenzare la neutralizzazione dell’anticorpo. Tuttavia, le mutazioni all’interno di queste regioni potrebbero influenzare la conformazione dell’RBD, l’attaccamento e la fusione, richiedendo ulteriori studi per determinare le loro conseguenze e il possibile effetto indiretto sull’evasione immunitaria. È stato scoperto che il D614G, ampiamente studiato, aumenta la capacità di RBD di spostarsi verso l’alto, necessaria per l’interazione con ACE2 [99].
Ciò ha comportato l’aumento dell’infettività e della trasmissibilità osservata per la variante D614G rispetto ai ceppi SARS-CoV-2 originali [100]. Il cambiamento di P681H è adiacente al sito di scissione della furina e potrebbe potenzialmente avere un effetto sulla scissione di S1/S2 e quindi sull’ingresso delle cellule e sull’infettività.
La variante di maggiore preoccupazione per quanto riguarda la fuga immunitaria, Beta o B.1.351, contiene sette mutazioni (D80A, D215G, K417N, E484K, N501Y, D614G, A701V) e tre delezioni (241/242/243del) nella proteina spike [75] (Figura 3). Due mutazioni (D80A, D215G) e tre residui deleti sono nel dominio N-terminale di S1, uno (A701V) è nel ciclo 2 di S2 e 3 sono nei residui chiave nel RBD (K417N, E484K, N501Y).
Finora, ci sono più studi che dimostrano che B.1.351 diminuisce la capacità di neutralizzazione degli anticorpi provocati dall’infezione con varianti precedenti o vaccinazione [48,83,101,102,103,104,105]. Questa riduzione del potenziale neutralizzante per B.1.351 viene rilevata più frequentemente in individui con bassi livelli di anticorpi e sta diminuendo più rapidamente con il tempo [105], aumentando le preoccupazioni sulla reinfezione o la protezione non ottimale da parte dei vaccini attuali.
Il problema nella popolazione non vaccinata esiste perché la maggior parte delle persone infette da SARS-CoV-2 sviluppa solo titoli da bassi a moderati, mentre titoli più alti si osservano solo in individui ospedalizzati gravemente. La perdita di attività neutralizzante del plasma convalescente contro B.1.351 variava da 11 a 33 volte e dai sieri dei vaccinati da 3,4 a 8,5 volte [50,83,101,103,104,105,106]. Inoltre, la variante B.1.351 ha mostrato resistenza alla neutralizzazione da parte della maggior parte dei mAb specifici per NTD e per un numero di mAb specifici per RBM [83,103,107].
La resistenza alla neutralizzazione anticorpale della variante B.1.351 è principalmente attribuita a tre mutazioni all’interno di RBD (K417N, E484K, N501Y). N501Y probabilmente non compromette la neutralizzazione da solo, ma piuttosto in combinazione con altri due, che sono stati trovati compromettere parzialmente la neutralizzazione generata da una precedente infezione o vaccinazione [48,103,106,107,108]. Il risultato del cambiamento in posizione 417 è la perdita dell’interazione polare con il residuo D30 sull’ACE2 umano [82].
Tuttavia, è stato dimostrato che una combinazione di K417N e N501Y migliora il legame con ACE2 e riduce il legame con gli anticorpi [109]. Questo miglioramento nel legame del recettore è supportato dall’osservazione di questa mutazione in un ceppo virulento adattato al topo di SARS-CoV-2 [110]. K417N ha dimostrato di essere cruciale per la fuga virale, abrogando efficacemente la neutralizzazione da parte di alcuni degli anticorpi neutralizzanti più comuni e potenti contro SARS-CoV-2 [103]. Contrariamente a ciò, altri [107] indicano che può contribuire alla neutralizzazione aumentando la probabilità di conversione alla conformazione aperta della proteina S, esponendo così gli epitopi alla neutralizzazione anticorpale.
La mutazione E484K, emersa indipendentemente in oltre 50 linee, corrisponde anche a un migliore legame con ACE2. Migliora ulteriormente l’affinità di legame di N501Y per ACE2, ma è stato associato anche alla fuga immunitaria da mAbs e sieri policlonali [48,49,83,106,107].
La sua posizione è all’interno della fessura di legame dell’RBD ed è considerato un epitopo neutralizzante dominante [75,108,111]. Il residuo 484 può mutare in una diversità di diversi amminoacidi (E484A, E484G, E448D ed E484K) sotto la pressione dei sieri convalescenti SARS-CoV-2 e mostra resistenza [112]. Si ritiene che l’impatto della mutazione 484 sull’evasione immunitaria sia significativamente aumentato dalla presenza di altre due mutazioni RBD in questa variante, ma è stato dimostrato anche il suo impatto come mutazione a punto singolo [106,112].
La variante B1.1.7 che porta la mutazione E484K è emersa ed è stata riconosciuta come una variante preoccupante nel Regno Unito e in Europa, poiché sembra essere responsabile di una significativa perdita aggiuntiva di capacità di neutralizzazione degli anticorpi monoclonali e policlonali [49].
È stato dimostrato che gli anticorpi monoclonali perdono quasi il 50% dell’attività neutralizzante contro B.1.1.7 che trasporta E484K. Una combinazione di E484K con varie mutazioni NTD (in particolare delezioni) potrebbe rivelarsi ancora più efficace nell’evasione immunitaria [113], che è la più significativa nei casi sia della variante Beta che di B1.1.7 con E484K.
Il terzo VOC riconosciuto a livello mondiale, Gamma o P.1, porta 11 mutazioni di picco. Cinque mutazioni si trovano all’interno di NTD (L18F, T20N, P26S, D138Y, R190S), tre in RBD (K417T, E484K, N501Y), due nel dominio C-terminale di S1 e vicino al sito di scissione della furina (D614G, H655Y), e uno in S2 (T1027I) (Figura 3). I sieri convalescenti e vaccinati mostrano una significativa perdita di attività neutralizzante contro P.1, ma la riduzione non è così sostanziale come contro B.1.315 [114,115,116].
La perdita di attività neutralizzante del plasma convalescente contro P.1 variava da 6,5 a 13 volte e dai sieri dei vaccinati da 2,2 a 2,8 volte [114,115], il che significa che la neutralizzazione di P.1 non era così gravemente compromessa come quella di B.1.351 e solo leggermente indebolito rispetto a quello di B.1.1.7. Non sorprende che l’attività di neutralizzazione di mAbs contro P.1 sia ridotta molto nello stesso modo di B.1.351, poiché le mutazioni triple RBD sono per lo più le stesse in entrambe le varianti [114].
La ragione delle differenze nella neutralizzazione di B.1.351 e P.1 da parte del siero immunitario riflette presumibilmente la differenza nelle mutazioni introdotte al di fuori del RBD. Il ruolo degli anticorpi neutralizzanti specifici per NTD non è ancora quasi definito. Si pensava che un’ampia schermatura glicanica legata all’N di NTD ne riducesse l’antigenicità, ma studi in vitro hanno mostrato la significativa capacità neutralizzante di alcuni anticorpi specifici per NTD [52].
Il fatto che NTD sia sotto pressione selettiva della risposta immunitaria umana è supportato dall’identificazione di delezioni di NTD in ospiti immunocompromessi con infezioni prolungate [79]. È possibile che i test di neutralizzazione basati su cellule bersaglio che sovraesprimono i recettori ACE2 siano responsabili della sottovalutazione del ruolo degli anticorpi specifici per NDT. Poiché i cambiamenti di NTD sono molto più distinti tra i tre principali VOC, sembra probabile che la variazione di neutralizzazione tra di loro sia dovuta piuttosto alle differenze di NTD rispetto a RBD.
Nel gennaio 2021, è stata segnalata l’emergere di una nuova variante in California portatrice di una mutazione L452R nel RBD [77]. Questa variante (Epsilon) comprende due linee separate B.1.427 e B.1.429, la prima portatrice di due mutazioni spike (L452R, D614G) e la seconda portatrice di quattro (S13I, W152C, L452R, D614G). Si presume che siano emersi già a maggio 2020 e abbiano ottenuto lo status di VOC negli Stati Uniti a causa del significativo aumento della frequenza da settembre 2020 a gennaio 2021. Nel febbraio 2021, sono stati identificati in >50% di tutti i casi sequenziati in California e molti altri stati [117].
È stato dimostrato che mostrano una moderata resistenza alla neutralizzazione da parte di sieri convalescenti (4-6,7 volte) e sieri di soggetti vaccinati (2-2,9 volte) [48,117]. La mutazione RBD L452R, condivisa da questi lignaggi, non si trova nella parte che interagisce direttamente con ACE2, ma si ipotizza che possa causare cambiamenti strutturali nella regione che promuovono l’interazione tra la proteina spike e il suo recettore ACE2.
Pertanto, l’infettività degli pseudovirus portatori di L452R è risultata superiore a quella della variante D614G ma leggermente ridotta rispetto a quella delle varianti N501Y [117]. Il meccanismo simile del cambiamento strutturale dell’RBD dovuto a L452R è offerto nella spiegazione della ridotta capacità di neutralizzazione degli anticorpi. Questa mutazione, tra molte altre mutazioni RBD, è stata selezionata da un pannello di anticorpi in vitro [112].
La variante emergente B.1.617 comprende tre sottolineaggi distinti (B.1.617.1, B.1.617.2, B.1.617.3) con diversi profili mutazionali [70]. Tuttavia, solo il sottolignaggio B.1.617.2 o Delta è ora riconosciuto a livello internazionale come VOC. È caratterizzato da mutazioni spike T19R, G142D, Δ157-158, L452R, T478K, D614G, P681R e D950N (Figura 3). Le altre due sottolinee hanno un profilo mutazionale simile: B.1.617.1 è definito dai cambiamenti di aminoacidi spike G142D, E154K, L452R, E484Q, D614G, P681R e Q1071H e B.1.617.3 è definito da T19R, L452R , E484Q, D614G, P681R e D950N. La presenza delle mutazioni RBD L452R, E484Q e D614G nel dominio C-terminale di S1 può comportare una maggiore trasmissibilità di questi sottolignaggi a causa del loro impatto noto sul legame ACE2 e sui cambiamenti conformazionali importanti per il legame ACE2.
Tutti e tre i sottolineaggi di B.1.617 mostrano P681R adiacente al sito di scissione della furina e hanno una maggiore scissione S da parte della furina, che si ipotizza aumenti la trasmissibilità e la patogenicità [118]. Sebbene il sottolignaggio B.1.617.2 fosse inizialmente considerato trasmissibile come B.1.1.7 [119], ulteriori prove dal Regno Unito, basate sulla probabilità che i contatti stretti di una persona infetta con la variante Delta diventino essi stessi infetti —il “tasso di attacco secondario”, suggerisce che questa variante potrebbe essere oltre il 60% più trasmissibile rispetto alla variante Alpha [120].
Secondo un recente rapporto, oltre il 90% dei nuovi casi di COVID-19 nel Regno Unito riguarda la variante Delta. La diffusione della variante Delta è registrata anche negli Stati Uniti, dove rappresenta ormai oltre il 6% di tutte le infezioni (più del 18% dei casi in alcuni stati occidentali degli USA) [121].
È previsto l’impatto sulla capacità di fuga immunitaria di tre sottolineaggi di B.1.617, a causa delle mutazioni RBD L452R, T478K ed E484Q e della loro combinazione con mutazioni e delezioni NTD, in particolare nel caso di B.1.617.2. Un cambiamento simile in posizione 478 (T478I) è stato precedentemente selezionato in vitro e ha mostrato di mostrare una neutralizzazione ridotta da parte di anticorpi monoclonali e sieri convalescenti umani [112].
Uno dei primi studi su B1.617.1 ha rivelato che la capacità di neutralizzazione dei sieri convalescenti e dei sieri dei destinatari del vaccino ucciso inattivato è stata mantenuta [122]. Altri studi hanno riportato una moderata riduzione della neutralizzazione di B1.617.1 da parte dei sieri di convalescenti e destinatari di vaccini mRNA e resistenza agli anticorpi monoclonali approvati per il trattamento COVID-19 [123,124,125]. È stato riscontrato che l’E484Q ha un impatto leggermente più lieve, ma corrisponde comunque all’effetto dell’E484K, che è una riduzione di 10 volte della neutralizzazione da parte dei sieri dei soggetti vaccinati. Inoltre, la combinazione di L452R ed E484Q non ha mostrato di avere un effetto additivo; piuttosto, la perdita di sensibilità era simile a quella osservata con ciascuna mutazione individualmente [124].
Infine, nella ricerca futura dovrebbe essere affrontato anche l’impatto delle varianti emergenti di SARS-CoV-2 preoccupanti sulla risposta immunitaria cellulare. È stato suggerito che la risoluzione dell’infezione da SARS-CoV-2 e di COVID-19 dipenda in modo significativo dalle risposte delle cellule T CD4+ e CD8+ [126], che svolgono anche un ruolo nella modulazione della gravità della malattia [45,127]. Negli individui convalescenti, l’immunità delle cellule T non è limitata agli epitopi derivati da spike e, quindi, sarebbe ragionevole presumere che rimarrebbe in gran parte intatta per le nuove varianti.
Tuttavia, nei destinatari della maggior parte dei vaccini attualmente disponibili, che offrono la proteina S come immunogeno bersaglio, l’immunità delle cellule T è limitata agli epitopi spike. Pertanto, è essenziale determinare se nuove mutazioni varianti in questi epitopi alterano le risposte delle cellule T in modo simile alla fuga dagli anticorpi neutralizzanti.
Tuttavia, gli studi che affrontano questo problema sono ancora scarsi, principalmente perché le misurazioni dell’immunità delle cellule T sono più impegnative per la pratica clinica di routine rispetto ai test di rilevamento degli anticorpi. Finora, l’effetto delle varianti B.1.1.7, B.1.351, P.1 e B.1.427/29 è risultato trascurabile sulle risposte delle cellule T sia CD4+ che CD8+ nei destinatari di vaccini a base di mRNA [ 128,129]. Ciò è stato supportato dal risultato di epitopi completamente conservati per il 93% delle cellule CD4+ e il 97% delle cellule T CD8+ nelle varianti.
Inoltre, è stato sottolineato che la capacità di legame dell’HLA non è influenzata nella maggior parte dei casi da una singola mutazione negli epitopi. Tuttavia, il repertorio di epitopi riconosciuti è probabilmente sostanzialmente diverso da un individuo all’altro a causa del polimorfismo HLA, e quindi l’impatto negativo delle mutazioni di specifiche varianti su ogni singola persona non può essere completamente ignorato [128].
L’incidenza dei lignaggi B.1.427/B.1.429 sta aumentando rapidamente
La variante SARS-CoV-2 B.1.427/B.1.429 è stata segnalata per la prima volta all’inizio del 2021 in California e a maggio 2021 è stata rilevata in 34 paesi aggiuntivi (41, 42). I due ceppi B.1.427 e B.1.429 (appartenenti al clade 20C secondo la designazione Nextstrain) condividono le stesse mutazioni S (S13I e W152C nel NTD e L452R nel RBD), ma ospitano mutazioni diverse in altri SARS-CoV- 2 geni (42).
L’analisi dell’orologio molecolare suggerisce che il capostipite di entrambi i lignaggi è emerso nel maggio 2020, divergendo per dare origine ai lignaggi B.1.427 e B.1.429 nel giugno-luglio 2020 (42). Il rapido aumento del numero di casi associati ai lignaggi B.1.427/B.1.429 ha portato alla loro classificazione come VOC dal Centro statunitense per il controllo delle malattie (https://www.cdc.gov/coronavirus/2019-ncov/ casi-aggiornamenti/variant-sorveglianza/variant-info.html).
Al 30 aprile 2021, in GISAID sono riportati 8.441 e 21.072 genomi sequenziati per i lignaggi B.1.427 e B.1.429, rispettivamente. Questo VOC è stato rilevato in California e in altri stati degli Stati Uniti e, più recentemente, in altri 34 paesi in tutto il mondo (Fig. 1, da A a H e tabella S1). Il numero di sequenze del genoma B.1.427/B.1.429 depositate è aumentato rapidamente dopo dicembre 2020, con un’incidenza superiore al 50% in California dal febbraio 2021 (Fig. 1, da B a E).
Nel complesso, questa analisi illustra l’aumento dell’incidenza del VOC B.1.427/B.1.429 e la sua progressiva diffusione geografica dalla California ad altri stati degli Stati Uniti e ad altri paesi, il che è coerente con un recente studio che suggerisce una maggiore trasmissibilità rispetto all’isolato ancestrale ( 42).

(A) Mappa mondiale che mostra la distribuzione geografica e i conteggi delle sequenze di B.1.427/B.1.429 VOC al 30 aprile 2021. (B) Conteggi cumulativi e individuali della sequenza B.1.427/B.1.429 di VOC per mese. (da C a E) Numero totale di sequenze SARS-CoV-2 (grigio) e B.1.427/B.1.429 VOC (blu/arancione) depositate mensilmente in tutto il mondo (C), negli Stati Uniti (D) e in California (E). (da F a H) Numero totale di sequenze B.1.427/B.1.429 (F), B.1.427 (G) e B.1.429 (H) depositate per paese al 30 aprile 2021. Solo paesi con n≥2 depositati vengono mostrate le sequenze
B.1.427/ B.1.429 S riduce la sensibilità agli Abs . provocati dal vaccino
Per valutare l’impatto delle tre mutazioni presenti nella glicoproteina B.1.427/B.1.429 S sulla neutralizzazione, abbiamo prima confrontato fianco a fianco la potenza di neutralizzazione degli Abs provocati dal vaccino mRNA contro G614 S e B.1.427/B. 1.429 Pseudovirus S. Abbiamo utilizzato il plasma di quindici individui che hanno ricevuto due dosi di vaccino Moderna mRNA-1273 e di quindici individui che hanno ricevuto due dosi di vaccino Pfizer/BioNtech BNT162b2 raccolti tra 7 e 27 giorni dopo l’immunizzazione di richiamo (tabella S2).
Tutti i vaccinati avevano una sostanziale attività di neutralizzazione del plasma contro i virus pseudotipizzati G614 SARS-CoV-2 S. Utilizzando un sistema di pseudotipizzazione del virus della leucemia murina (MLV), i titoli medi geometrici (GMT) hanno mostrato che la potenza di neutralizzazione media del plasma suscitato dall’mRNA1273 di Moderna è stata ridotta di 2,4 volte per B.1.427/B.1.429 S (GMT: 178) rispetto a G614 S (GMT: 424) (Fig. 2, A e B; Fig. S1 e S2; e tabella S3) mentre è stato ridotto di 2,3 volte con plasma indotto da Pfizer/BioNtech BNT162b2 (B.1.427/B.1.429 GMT: 78 contro G614 GMT: 182) (Fig. 2, C e D; figg. S1 e S2; e tabella S3).
Utilizzando un sistema di pseudotipizzazione del virus della stomatite vescicolare (VSV), abbiamo osservato una riduzione media di 2,2 volte dell’attività di neutralizzazione del plasma indotta dall’mRNA1273 di Moderna contro B.1.427/B.1.429 S (GMT: 213) rispetto a G614 S (GMT: 464) pseudovirus (Fig. 2, E e F; figg. S1 e S2; e tabella S3) e una riduzione media di 2,5 volte dell’attività neutralizzante del plasma indotta da Pfizer/BioNtech BNT162b2 contro B.1.427/B.1.429 S (GMT: 113 ) rispetto agli pseudovirus G614 S (GMT: 285) (Fig. 2, G e H; figg. S1 e S2; e tabella S3).
Abbiamo anche analizzato il plasma di 18 individui, 5 dei quali erano stati precedentemente infettati da SARS-CoV-2 di tipo selvaggio, che hanno ricevuto due dosi di vaccino Pfizer/BioNtech BNT162b2 e i cui campioni sono stati raccolti tra 14 e 28 giorni dopo l’immunizzazione di richiamo. Abbiamo confrontato la potenza di neutralizzazione degli Abs indotti dal vaccino Pfizer/BioNtech BNT162b2 contro D614 S, B.1.427/B.1.429 S, B.1.1.7 S, B.1.351 S e P.1 S VSV pseudotipizzati virus che esprimono Vero E6 TMPRSS2 come cellule bersaglio.
La potenza di neutralizzazione plasmatica dei GMT è stata ridotta di 2,9 volte per B.1.427/B.1.429 S (GMT: 197) rispetto a D614 S (GMT: 570), che è una diminuzione paragonabile a quella osservata con B.1.351 (GMT: 180, riduzione di 3,2 volte) e maggiore di quella osservata con virus pseudotipizzati B.1.1.7 e P.1 (GMT: 450 e 330, riduzione di 1,3 volte e 1,7 volte) (Fig. 2, I e J; figg. S1 e S2 e tabella S3). Questi dati indicano che le tre sostituzioni di residui B.1.427/B.1.429 S portano a una riduzione modesta ma significativa della potenza di neutralizzazione degli Abs indotti dal vaccino.

(A, B, E e F) Titoli Ab neutralizzanti (ID50) mostrati come connessi a coppie [(A) e (E)] o il titolo della media geometrica, GMT [(B) e (F)] rispetto a MLV [(A) ) e (B)] o virus pseudotipizzati VSV [(E) e (F)] che ospitano G614 SARS-CoV-2 S o B.1.427/B.1.429 (B.1.429) S determinati utilizzando plasma di individui che hanno ricevuto due dosi del vaccino Moderna mRNA-1273 (blu). (C, D, G e H) Titoli Ab neutralizzanti (ID50) mostrati come connessi a coppie [(C) e (G)] o il titolo della media geometrica, GMT [(D) e (H)] rispetto a MLV [(C) ) e (D)] o virus pseudotipizzati VSV [(G) e (H)] che ospitano G614 SARS-CoV-2 S o B.1.427/B.1.429 (B.1.429) S determinati utilizzando plasma di individui che hanno ricevuto due dosi del vaccino mRNA Pfizer/BioNtech BNT162b2 (rosso). (I e J) Neutralizzazione dei titoli Ab ID50 (I) e GMT (J) contro virus pseudotipizzati VSV che ospitano D614 SARS-CoV-2 S, B.1. 427/B.1.429 S, B.1.1.7 S, B.1.351 S o P.1 S determinati utilizzando plasma da individui naïve (blu) e precedentemente infetti (rosso) che hanno ricevuto due dosi di vaccino Pfizer/BioNtech BNT162b2 mRNA . Naïve: individui vaccinati che non erano stati precedentemente infettati da SARS-CoV-2. Immune: individui vaccinati che erano stati precedentemente infettati da SARS-CoV-2. (K e L) Neutralizzazione dei titoli Ab ID50 (K) e GMT (L) contro virus pseudotipizzati VSV che ospitano D614 SARS-CoV-2 S, B.1.427/B.1.429 S, B.1.1.7 S, B.1.351 S o P.1 S determinato utilizzando plasma di individui convalescenti infettati da SARS-CoV-2 di tipo selvatico. I dati di neutralizzazione mostrati da (A) a (H) e da (I) a (L) sono stati eseguiti utilizzando rispettivamente 293T-ACE2 e VeroE6-TMPRSS2. I dati sono una media di n = 2 repliche. 1 S determinato utilizzando il plasma di individui naïve (blu) e precedentemente infetti (rosso) che hanno ricevuto due dosi di vaccino mRNA Pfizer/BioNtech BNT162b2. Naïve: individui vaccinati che non erano stati precedentemente infettati da SARS-CoV-2. Immune: individui vaccinati che erano stati precedentemente infettati da SARS-CoV-2. (K e L) Neutralizzazione dei titoli Ab ID50 (K) e GMT (L) contro virus pseudotipizzati VSV che ospitano D614 SARS-CoV-2 S, B.1.427/B.1.429 S, B.1.1.7 S, B.1.351 S o P.1 S determinato utilizzando plasma di individui convalescenti infettati da SARS-CoV-2 di tipo selvatico. I dati di neutralizzazione mostrati da (A) a (H) e da (I) a (L) sono stati eseguiti utilizzando rispettivamente 293T-ACE2 e VeroE6-TMPRSS2. I dati sono una media di n = 2 repliche. 1 S determinato utilizzando plasma di individui naïve (blu) e precedentemente infetti (rosso) che hanno ricevuto due dosi di vaccino mRNA Pfizer/BioNtech BNT162b2. Naïve: individui vaccinati che non erano stati precedentemente infettati da SARS-CoV-2. Immune: individui vaccinati che erano stati precedentemente infettati da SARS-CoV-2. (K e L) Neutralizzazione dei titoli Ab ID50 (K) e GMT (L) contro virus pseudotipizzati VSV che ospitano D614 SARS-CoV-2 S, B.1.427/B.1.429 S, B.1.1.7 S, B.1.351 S o P.1 S determinato utilizzando plasma di individui convalescenti infettati da SARS-CoV-2 di tipo selvatico. I dati di neutralizzazione mostrati da (A) a (H) e da (I) a (L) sono stati eseguiti utilizzando rispettivamente 293T-ACE2 e VeroE6-TMPRSS2. I dati sono una media di n = 2 repliche. individui vaccinati che non erano stati precedentemente infettati da SARS-CoV-2. Immune: individui vaccinati che erano stati precedentemente infettati da SARS-CoV-2. (K e L) Neutralizzazione dei titoli Ab ID50 (K) e GMT (L) contro virus pseudotipizzati VSV che ospitano D614 SARS-CoV-2 S, B.1.427/B.1.429 S, B.1.1.7 S, B.1.351 S o P.1 S determinato utilizzando plasma di individui convalescenti infettati da SARS-CoV-2 di tipo selvatico. I dati di neutralizzazione mostrati da (A) a (H) e da (I) a (L) sono stati eseguiti utilizzando rispettivamente 293T-ACE2 e VeroE6-TMPRSS2. I dati sono una media di n = 2 repliche. individui vaccinati che non erano stati precedentemente infettati da SARS-CoV-2. Immune: individui vaccinati che erano stati precedentemente infettati da SARS-CoV-2. (K e L) Neutralizzazione dei titoli Ab ID50 (K) e GMT (L) contro virus pseudotipizzati VSV che ospitano D614 SARS-CoV-2 S, B.1.427/B.1.429 S, B.1.1.7 S, B.1.351 S o P.1 S determinato utilizzando plasma di individui convalescenti infettati da SARS-CoV-2 di tipo selvatico. I dati di neutralizzazione mostrati da (A) a (H) e da (I) a (L) sono stati eseguiti utilizzando rispettivamente 293T-ACE2 e VeroE6-TMPRSS2. I dati sono una media di n = 2 repliche. 429 S, B.1.1.7 S, B.1.351 S o P.1 S determinati utilizzando plasma di individui convalescenti infettati da SARS-CoV-2 di tipo selvatico. I dati di neutralizzazione mostrati da (A) a (H) e da (I) a (L) sono stati eseguiti utilizzando rispettivamente 293T-ACE2 e VeroE6-TMPRSS2. I dati sono una media di n = 2 repliche. 429 S, B.1.1.7 S, B.1.351 S o P.1 S determinati utilizzando plasma di individui convalescenti infettati da SARS-CoV-2 di tipo selvatico. I dati di neutralizzazione mostrati da (A) a (H) e da (I) a (L) sono stati eseguiti utilizzando rispettivamente 293T-ACE2 e VeroE6-TMPRSS2. I dati sono una media di n = 2 repliche.
Abbiamo anche analizzato il plasma di 9 donatori convalescenti, che hanno manifestato COVID-19 sintomatico all’inizio del 2020 (e di conseguenza sono stati probabilmente esposti al Wuhan-1 o a un isolato SARS-CoV-2) strettamente correlato, raccolto da 15 a 28 giorni dopo l’insorgenza dei sintomi (tabella S2). La potenza di neutralizzazione del plasma di 9 donatori convalescenti è stata ridotta di 3,4 volte per B.1.427/B.1.429 S (GMT: 70) rispetto a G614 S (GMT: 240), simile a quanto osservato con B.1.351 (4.4- volte, GMT: 55) e P.1 (3,3 volte, GMT: 72) virus pseudotipizzati, mentre la neutralizzazione di B.1.1.7 è stata meno colpita (1,9 volte, GMT: 127) (Fig. 2, K e L , figg. S1 e S2 e tabella S3). In diversi casi il livello di attività neutralizzante contro i VOC è risultato inferiore al limite di rilevabilità.
Questi risultati mostrano che le tre mutazioni presenti nella glicoproteina B1.427/B.1.429 S diminuiscono l’attività neutralizzante degli Ab indotti dal vaccino e dall’infezione, suggerendo che queste sostituzioni di residui che definiscono il lignaggio sono associate all’evasione immunitaria. Tuttavia, questi dati sottolineano anche la maggiore qualità delle risposte Ab indotte dalla vaccinazione rispetto all’infezione e la loro maggiore resilienza alle mutazioni trovate nei VOC.
B.1.427/B.1.429 Le mutazioni S riducono la sensibilità agli Abs specifici per RBD e NTD
Per valutare il contributo delle sostituzioni RBD e NTD alla ridotta potenza di neutralizzazione dei sieri di vaccinati e plasma convalescente, abbiamo confrontato l’attività neutralizzante di 34 RBD e 10 NTD mAbs contro la variante D614 S o B.1.427/B.1.429 S utilizzando un Sistema di pseudotipizzazione VSV (1, 43).
Il pannello di mAb RBD-specifici (inclusi 6 mAb clinici) riconosce siti antigenici distinti come precedentemente caratterizzati (10, 11, 13, 20, 44, 45). In breve, gli epitopi abbracciano l’RBM (siti antigenici Ia e Ib), un sito antigenico criptico II, il sito antigenico IV contenente glicano N343 esposto e un secondo sito antigenico criptico V (10, 11).
Un totale di 14 su 34 mAb ha mostrato una potenza di neutralizzazione ridotta quando si confrontano gli pseudovirus B.1.427/B.1.429 S e D614 S (Fig. 3, da A a C e fig. S3). Considerando i 6 mAb in uso clinico: regdanvimab (CT-P59), e in misura minore etesvimab (LY-CoV016), hanno mostrato una riduzione della potenza di neutralizzazione, mentre bamlanivimab (LY-CoV555) ha perso completamente la sua attività neutralizzante.
La neutralizzazione mediata dal cocktail di mAb casirivimab/imdevimab (REGN10933 e REGN10987) (14, 15), e da VIR-7831 (derivato di S309, recentemente ribattezzato sotrovimab) (10, 23, 24), non è influenzata dal B.1.427/ B.1.429 variante S. Per affrontare il ruolo della mutazione L452R nella fuga di neutralizzazione da Abs RBD-specifici, abbiamo testato il legame dei 34 mAb RBD-specifici a WT e RBD mutante L452R mediante interferometria a biostrato (fig. S4). I 10 mAb specifici per RBD che sperimentano una riduzione di 10 volte o maggiore della potenza di neutralizzazione della variante B.1.427/B.1.429, rispetto a D614 S, si sono legati scarsamente al mutante L452R RBD, dimostrando un ruolo diretto di questa mutazione im immune evasione.

(A e D) Neutralizzazione di VSV pseudotipizzato SARS-CoV-2 che trasporta la proteina S D614 (grigio) o B.1.427/B.1.429 (arancione) mediante mAb RBD in fase clinica (A) e un mAb con target NTD (S2X333) (D). I dati sono rappresentativi di n = 2 repliche. (B ed E) Neutralizzazione degli pseudotipi SARS-CoV-2 S VSV portatori di D614 o B.1.427/B.1.429 S da 34 mAb diretti all’RBD e 10 mAb diretti a NTD. I dati sono la media dei valori di concentrazione inibitoria (IC50) del 50% (ng/ml) di n = 2 esperimenti indipendenti. I titoli di IC50 non neutralizzanti sono stati fissati a 105 ng/ml. (C e F) La neutralizzazione da mAb specifici per RBD (C) e NTD (G) ha mostrato come valori medi di IC50 (in alto) e variazione media di piega (in basso) per B.1.427/B.1.429 S (arancione) rispetto a D614G S (grigio) Pseudovirus VSV. VIR-7831 è un derivato di S309 mAb (sotrovimab). *, VIR-7832 (variante di VIR-7831 che porta le mutazioni LS-GAALIE Fc) mostrato come quadrati. I titoli di IC50 non neutralizzanti e la variazione di piegatura sono stati fissati rispettivamente a 105 ng/ml e 104.
Abbiamo scoperto che l’attività neutralizzante di tutti i 10 mAb specifici per NTD testati è stata abolita a causa della presenza delle mutazioni S13I e W152C (Fig. 3, da D a F). Questi dati indicano che la ridotta potenza di neutralizzazione della variante B.1.427/B.1.429 deriva dall’evasione della neutralizzazione mediata da mAb sia specifica per RBD che per NTD.
Caratterizzazione strutturale del trimero SARS-CoV-2 B.1.427/B.1.429 S
Per visualizzare i cambiamenti in SARS-CoV-2 B.1.427/B.1.429 S che contribuiscono all’evasione immunitaria, abbiamo determinato una struttura cryoEM del trimero ectodomain variante S (portatore delle mutazioni HexaPro (46)) legato al trimero specifico per RBD mAb S2M11 e mAb S2L20 specifico per NTD con risoluzione di 2,3 (Fig. 4A, fig. S5 e tabella S4). S2M11 è stato utilizzato per bloccare gli RBD nello stato chiuso (Fig. 4B) mentre S2L20 è stato utilizzato per stabilizzare i NTD (Fig. 4C) (12, 13). La sovrapposizione delle strutture RBD SARS-CoV-2 legate a regdanvimab- (CT-P59) e bamlanivimab- (LY-CoV555) a B.1.427/B.1.429 S rivela che l’L452R introdotto è stericamente incompatibile con il legame di questi mAbs (Fig. 4, D ed E), razionalizzando lo smorzamento o la perdita dell’attività neutralizzante.

(A) Struttura del trimero S (rendering superficiale) legato ai Fab S2M11 e S2L20 (nastri) in due orientamenti ortogonali. I protomeri SARS-CoV-2 S sono di colore rosa, ciano e oro, mentre le catene pesanti e leggere S2L20 Fab sono rispettivamente di colore scuro e verde chiaro e le catene pesanti e leggere S2M11 Fab sono rispettivamente di colore scuro e grigio chiaro. Nella mappa vengono risolti solo i domini delle variabili Fab. I glicani N-linked sono resi come sfere blu scuro. (B) Ingrandito nella vista dell’RBD associato a S2M11 con R452 mostrato nella rappresentazione della palla e del bastone. (C) Ingrandito in vista del NTD legato a S2L20 con terminale N disordinato, supersito regions-tornante e regioni ad anello mostrate come linee tratteggiate. (D) Sovrapposizione della struttura RBD SARS-CoV-2 legata a CT-P59 (PDB 7CM4) su SARS-CoV-2 B.1.427/B.1. La struttura 429 S cryoEM mostra che R452 si scontra stericamente con il mAb. (E) La sovrapposizione della struttura SARS-CoV-2 RBD legata a LY-CoV555 (PDB 7KMG) sulla struttura crioEM SARS-CoV-2 B.1.427/B.1.429 S mostra che L452R si scontra stericamente con il mAb. (F) La sovrapposizione della struttura SARS-CoV-2 S legata a S2X333 (PDB 7LXW) sulla struttura crioEM SARS-CoV-2 B.1.427/B.1.429 S rivela che la maggior parte dei residui di epitopi antigenici del supersito NTD sono disordinati. (G) La sovrapposizione della struttura RBD SARS-CoV-2 legata ad ACE2 (PDB 7DMU) sulla struttura CryoEM SARS-CoV-2 B.1.427/B.1.429 S mostra che L452R punta lontano dall’interfaccia con ACE2 (F) La sovrapposizione della struttura SARS-CoV-2 S legata a S2X333 (PDB 7LXW) sulla struttura crioEM SARS-CoV-2 B.1.427/B.1.429 S rivela che la maggior parte dei residui di epitopi antigenici del supersito NTD sono disordinati. (G) La sovrapposizione della struttura RBD SARS-CoV-2 legata ad ACE2 (PDB 7DMU) sulla struttura CryoEM SARS-CoV-2 B.1.427/B.1.429 S mostra che L452R punta lontano dall’interfaccia con ACE2 (F) La sovrapposizione della struttura SARS-CoV-2 S legata a S2X333 (PDB 7LXW) sulla struttura crioEM SARS-CoV-2 B.1.427/B.1.429 S rivela che la maggior parte dei residui di epitopi antigenici del supersito NTD sono disordinati. (G) La sovrapposizione della struttura RBD SARS-CoV-2 legata ad ACE2 (PDB 7DMU) sulla struttura crioEM SARS-CoV-2 B.1.427/B.1.429 S mostra che L452R punta lontano dall’interfaccia con ACE2
Successivamente abbiamo utilizzato il raffinamento locale per tenere conto delle dinamiche conformazionali di NTD e S2L20 rispetto al resto di S e abbiamo ottenuto una ricostruzione cryoEM del NTD legato a S2L20 a una risoluzione di 3.0 (Fig. 4C, fig. S5 e tabella S4) . La struttura rivela che il supersito antigenico B.1.427/B.1.429 NTD è gravemente alterato. Il capolinea N è disordinato fino al residuo 27, così come il tornante β del supersito (disordinato tra i residui 137-158) e l’ansa del supersito (disordinato tra i residui 243-264) (Fig. 4F). Questi cambiamenti strutturali spiegano l’abrogazione del legame e la neutralizzazione del pannello di mAb specifici per NTD valutato.
La sovrapposizione di una struttura RBD SARS-CoV-2 legata ad ACE2 con la struttura S variante B.1.427/B.1.429 mostra che il residuo R452 punta lontano e non contatta ACE2, suggerendo che questa sostituzione non influenzerebbe l’impegno del recettore (Fig. 4G). Successivamente abbiamo valutato il legame dell’ectodominio umano ACE2 monomerico a B.1.427/B.1.429 immobilizzato e RBD di tipo selvatico utilizzando l’interferometria del biostrato di risonanza plasmonica di superficie (fig. S6, A e B e tabella S5) (fig. S6, da C a E, e tabella S5) nonché il legame di B.1.427/B.1.429, B.1.1.7 e RBD wild-type all’ACE2 umano immobilizzato mediante ELISA (fig. S6F e tabella SS5). I nostri risultati indicano che i RBD B.1.427/B.1.429 e wildtype si sono legati ad ACE2 con affinità comparabili (mentre il B.1.1.7 RBD aveva un’affinità notevolmente aumentata per ACE2 (34)), convalidando le osservazioni strutturali.
Riarrangiamento del legame disolfuro nella variante B.1.427/B.1.429 del supersito antigenico NTD
Per indagare ulteriormente sulle basi molecolari della perdita dell’attività di neutralizzazione del mAb diretta da NTD e dei cambiamenti strutturali nel NTD, abbiamo analizzato il legame di un pannello di mAb specifici per NTD alle varianti NTD SARS-CoV-2 ricombinanti utilizzando ELISA. La mutazione del peptide del segnale S13I ha attenuato il legame di 5 mAb e ha abrogato il legame di 5 mAb aggiuntivi su 11 mAb neutralizzanti valutati (Fig. 5A e fig. S7).
Inoltre, la mutazione W152C ha ridotto il riconoscimento di 6 mAb neutralizzanti NTD, inclusa una completa perdita di legame per due di essi, con un pattern complementare a quello osservato per S13I (Fig. 5A e fig. S7). L’NTD B.1.427/B.1.429 S13I/W152C non si è legato ad alcun mAb neutralizzante diretto da NTD, che è noto per colpire un singolo sito antigenico (sito antigenico i) (12), mentre il legame del mAb S2L20 non neutralizzante al sito antigenico NTD iv non è stato influenzato da alcun mutante, confermando la corretta ritenzione del ripiegamento, come supportato dai dati strutturali (Fig. 5A e fig. S7).
Il legame del plasma indotto dal vaccino ai mutanti NTD ha confermato ed esteso queste osservazioni con Abs policlonali, mostrando una riduzione sempre più marcata dei titoli di legame a causa delle sostituzioni dei residui W152C, S13I e S13I/W152C (Fig. 5B e fig. S8).

(A) Legame di un pannello di 11 mAb neutralizzanti (sito antigenico i) e 1 non neutralizzante (sito antigenico iv) specifici per NTD a varianti NTD ricombinanti di SARS-CoV-2 analizzate da ELISA visualizzate come una mappa di calore. (B) Legame di plasma Abs da individui vaccinati a varianti NTD SARS-CoV-2 ricombinanti analizzate da ELISA. Il fattore di diluizione medio per ciascun mutante è stato confrontato con il test ANOVA unidirezionale con valori di p wildtype <0.05 ( ) e <0.001 (*). (da C a G) Spettri di massa deconvoluti di costrutti NTD purificati, compreso il NTD di tipo selvatico con il peptide segnale nativo (B), il NTD S13I (C), il NTD S13I e W152C (D), il NTD W152C (E) e l’S12F NTD (F). La massa empirica (nero) e la massa teorica (rosso) sono mostrate accanto al picco corrispondente. Ulteriori 119 Da sono stati osservati per i NTD S13I e W152C corrispondenti alla cisteinilazione del residuo di cisteina libera in questi costrutti (poiché la L-cisteina era presente nei mezzi di espressione). Il peptide segnale scisso (testo blu) e la successiva sequenza di residui (testo nero) sono mostrati anche in base ai risultati MS. I residui mutati sono mostrati in grassetto. Le cisteine sono evidenziate in arancione chiaro (a meno che nel peptide segnale scisso) mentre i legami disolfuro sono mostrati come linee punteggiate arancione chiaro tra le cisteine.
Abbiamo precedentemente dimostrato che la rottura del legame disolfuro C15/C136 che collega il terminale N al resto del NTD, attraverso la mutazione del residuo o l’alterazione del sito di scissione del peptide segnale, abroga l’attività neutralizzante degli mAb che prendono di mira il supersito antigenico NTD ( sito i) (12).
Poiché la sostituzione S13I risiede nel peptide segnale e si prevede che sposti il sito di scissione del peptide segnale da S13-Q14 a C15-V16, abbiamo ipotizzato che questa sostituzione influenzi indirettamente l’integrità del sito antigenico NTD i, che comprende il terminale N. L’analisi della spettrometria di massa delle varianti NTD S13I e S13I/W152C ha confermato che la scissione del peptide segnale avviene immediatamente dopo il residuo C15 (Fig. 5, da C a E).
Di conseguenza, C136, che altrimenti sarebbe disolfuro legato a C15, è cisteinilato nell’S13I NTD a causa della presenza di cisteina libera nei mezzi di espressione (Fig. 5D e fig. S9). Allo stesso modo, anche la mutazione W152C, che introduce una cisteina libera, è risultata cisteinilata nel NTD W152C (Fig. 5E). Non è chiaro se la cisteinilazione si verifichi durante l’infezione naturale con i soli mutanti S13I o W152C, o quale contributo la cisteinilazione giochi nell’evasione immunitaria dei soli mutanti S13I o W152C. In particolare, lo smorzamento del legame mAb neutralizzante specifico per NTD è più forte per il mutante S13I rispetto al mutante S12P che in precedenza abbiamo mostrato sposta anche il sito di scissione del peptide segnale a C15-V16 (Fig. 5A).
Al contrario, non abbiamo osservato alcun effetto sul legame con mAb della sostituzione S12F, che è stata rilevata anche in isolati clinici, in accordo con il fatto che questa mutazione non ha influenzato il sito di scissione del peptide segnale nativo (cioè, si verifica al S13 -Q14 posizione), come osservato dalla spettrometria di massa (Fig. 5G). In assenza del legame disolfuro C15-C136 l’N terminale non è più graffato al NTD, coerentemente con i dati strutturali che mostrano che l’N terminale della variante B.1.427/B.1.429 diventa disordinato rispetto al resto del NTD (Fig. 4C).
Sebbene le varianti NTD S13I e W152C fossero rispettivamente cisteinilate nelle posizioni C136 e W152C, il doppio mutante S13I/W152C non era cisteinilato, suggerendo che C136 e W152C avevano formato un nuovo legame disolfuro. (Fig. 5, da D a F). L’analisi di spettrometria di massa tandem di peptidi non ridotti e digeriti ha identificato peptidi discontinui collegati contenenti C136 e W152C (fig. S9) confermando che si forma un legame disolfuro tra C136 e W152C nel NTD S13I/W152C del B.1.427/B.1.429 variante. W152C si trova nel tornante del supersito antigenico e la formazione di un nuovo legame disolfuro con C136 sposterebbe i residui nel tornante >20 e la struttura locale del tornante era disordinata nel B.1.427/ B.1.429 variante (Fig. 4C).
Collettivamente, questi risultati dimostrano che le mutazioni S13I e W152C trovate nella variante B.1.427/B.1.429 S sono congiuntamente responsabili della fuga da mAbs specifici per NTD, a causa della delezione dei due residui N-terminali di SARS-CoV-2 S. e riarrangiamento generale del supersito antigenico NTD. I nostri dati supportano che il NTD SARS-CoV-2 ha evoluto un meccanismo compensatorio per formare un legame disolfuro alternativo e che le mutazioni del peptide segnale S si verificano in vivo in un contesto clinico per promuovere l’evasione immunitaria. La variante SARS-CoV-2 B.1.427/B.1.429 S si basa quindi su una strategia di fuga di neutralizzazione indiretta e insolita.
link di riferimento: https://science.sciencemag.org/content/early/2021/06/30/science.abi7994
link di riferimento: https://www.mdpi.com/1999-4915/13/7/1192/htm



{kind=link}