










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Ricercatori israeliani dell’Università di Tel Aviv hanno identificato con successo 3 proteine chiave su un totale di 29 proteine trovate nel genoma SAR-CoV-2 che possono danneggiare i vasi sanguigni dell’ospite umano. Queste tre proteine chiave erano nsp2, nsp5_c145a (mutante catalitico morto di nsp5) e nsp7.
Ad oggi è noto che il genoma della SARS-CoV-2 codifica per 29 proteine, il cui contributo alle manifestazioni della malattia, e soprattutto delle complicanze endoteliali, è sconosciuto.
Il team di studio ha clonato ed espresso 26 di queste proteine nelle cellule umane e ha caratterizzato la risposta endoteliale alla sovraespressione di ciascuna, individualmente.
I ricercatori hanno dimostrato che mentre la maggior parte delle proteine SARS-CoV-2 induceva cambiamenti significativi nella permeabilità endoteliale, nsp2, nsp5_c145a (mutante catalitico morto di nsp5) e nsp7 riducevano anche il CD31 e aumentavano l’espressione del fattore von Willebrand e IL-6, suggerendo una disfunzione endoteliale. .
Utilizzando l’analisi basata sulla propagazione di una rete di interazione proteina-proteina (PPI), il team di studio ha previsto le proteine endoteliali interessate dalle proteine virali che potenzialmente mediano questi effetti.
Il team ha ulteriormente applicato il modello PPI per identificare il ruolo di ciascuna proteina SARS-CoV-2 in altri tessuti colpiti dalla malattia da coronavirus (COVID-19).
Durante la convalida del modello di rete PPI, il team di studio ha scoperto che le proteine a giunzione stretta (TJ) caderina-5, ZO-1 e β-catenina sono influenzate da nsp2, nsp5_c145a e nsp7 coerentemente con la previsione del modello.
I risultati dello studio identificano le proteine SARS-CoV-2 che potrebbero essere più dannose in termini di disfunzione endoteliale, facendo così luce sugli aspetti vascolari di COVID-19.
I risultati dello studio sono stati pubblicati nella rivista peer reviewed: eLIfe
https://elifesciences.org/articles/69314
La malattia da coronavirus (COVID-19) causata dal nuovo coronavirus del 2019 (2019-nCoV/SARS-CoV-2) ha portato a una pandemia globale nel 2020. Alla fine di settembre 2021, il coronavirus aveva infettato oltre 220 milioni di persone in tutto il mondo, causando oltre 4,5 milioni di morti.
Dopo la fase iniziale dell’infezione virale, circa il 30% dei pazienti ospedalizzati con COVID-19 sviluppa una malattia grave con danno polmonare progressivo, nota come sindrome respiratoria acuta grave (SARS), e una grave risposta immunitaria. È interessante notare che sono state osservate ulteriori patologie, come ipossiemia e tempesta di citochine che, in alcuni casi, portano a insufficienza cardiaca e renale e sintomi neurologici.
Osservazioni recenti suggeriscono che queste patologie siano dovute principalmente all’aumento della coagulazione e alla disfunzione vascolare (Lee et al., 2021; Libby e Lüscher, 2020; Siddiqi et al., 2020). Attualmente si ritiene che oltre ad essere una malattia respiratoria, il COVID-19 possa anche essere una “malattia vascolare” (Lee et al., 2021), poiché potrebbe causare una barriera vascolare che perde e una maggiore espressione del fattore di von Willebrand ( VWF) (Siddiqi et al., 2020), responsabile dell’aumento della coagulazione, del rilascio di citochine e dell’infiammazione (Siddiqi et al., 2020; Teuwen et al., 2020; Aid et al., 2020; Potus et al., 2020; Wazny et al., 2020; Pum et al., 2021; Barbosa et al., 2021; Lin et al., 2020; Matarese et al., 2020; Xiao et al., 2020).
Studi recenti suggeriscono che il principale meccanismo di interruzione della barriera endoteliale si verifica in più fasi: in primo luogo, un effetto diretto sulle cellule endoteliali che provoca una risposta immunitaria dell’endotelio vascolare (endoteliite) e disfunzione endoteliale.
Secondo, lisi e morte delle cellule endoteliali Teuwen et al., 2020; Xiao et al., 2020 seguito dal sequestro dell’enzima di conversione dell’angiotensina I umana 2 (hACE2) da parte delle proteine virali spike che attivano le vie callicreina-bradichinina e renina-angiotensina, aumentando la permeabilità vascolare (Teuwen et al., 2020; Varga et al. ., 2020).
Infine, la reazione eccessiva del sistema immunitario, durante la quale una combinazione di neutrofili e cellule immunitarie che producono specie reattive dell’ossigeno, citochine infiammatorie (p. es., interleuchina [IL]-1β, IL-6 e fattore di necrosi tumorale) e molecole vasoattive (p. es., trombina, istamina, trombossano A2 e fattore di crescita endoteliale vascolare) e la deposizione di acido ialuronico portano alla rottura delle giunzioni endoteliali, aumento della permeabilità vascolare e perdite e coagulazione (Libby e Lüscher, 2020; Teuwen et al., 2020; Varga et al., 2020).
Di grande interesse è l’effetto sul sistema vascolare del cervello. È stato suggerito che gli effetti cerebrovascolari siano tra gli effetti di lunga durata di COVID-19. In effetti, è stato scoperto che la suscettibilità delle cellule endoteliali cerebrali all’infezione diretta da SARS-CoV-2 aumenta a causa dell’aumentata espressione di hACE2 in modo dipendente dal flusso sanguigno, portando a un processo di espressione genica unico che potrebbe contribuire agli effetti cerebrovascolari del virus (Pober e Sessa, 2007).
Mentre molti studi sottolineano l’importanza del sistema vascolare nel COVID-19 (Kaneko et al., 2021; Jung et al., 2020b; Nägele et al., 2020), solo pochi Pons et al., 2020; Chioh et al., 2020; Nascimento Conde et al., 2020; Buzhdygan et al., 2020 hanno esaminato la risposta vascolare diretta al virus.
La maggior parte di questi rapporti deriva da osservazioni cliniche, studi in vitro o studi in vivo in cui animali/cellule sono stati trasfettati con il virus SARS-CoV-2 e la loro risposta cellulare sistemica è stata valutata, senza individuare le proteine virali specifiche che causano i cambiamenti osservati. SARS-CoV-2 è un virus avvolto con un genoma di RNA a singolo filamento a senso positivo di 30 kb, che codifica per 29 proteine (Figura 1).
Queste proteine possono essere classificate come: proteine strutturali : S (proteine spike), E (proteine dell’involucro), M (proteine di membrana), N (proteina nucleocapside e RNA virale); proteine non strutturali : nsp1–16; proteine accessorie della cornice di lettura aperta : orf3-10 (Kim et al., 2020; Hu et al., 2021). La tabella 1 riassume gli effetti noti di specifiche proteine SARS-CoV-2 (Gordon et al., 2020; Peng et al., 2020b; Procko, 2020; Cornillez-Ty et al., 2009; Romano et al., 2020; Hillen et al., 2020; Chi et al., 2003).
La funzionalità di alcuni di questi è ancora sconosciuta. Inoltre, esiste ancora una notevole lacuna nelle conoscenze sui meccanismi molecolari, in particolare sui percorsi di interazione proteina-proteina (PPI) (Cowen et al., 2017), che portano alla disfunzione dei tessuti.
Figura 1
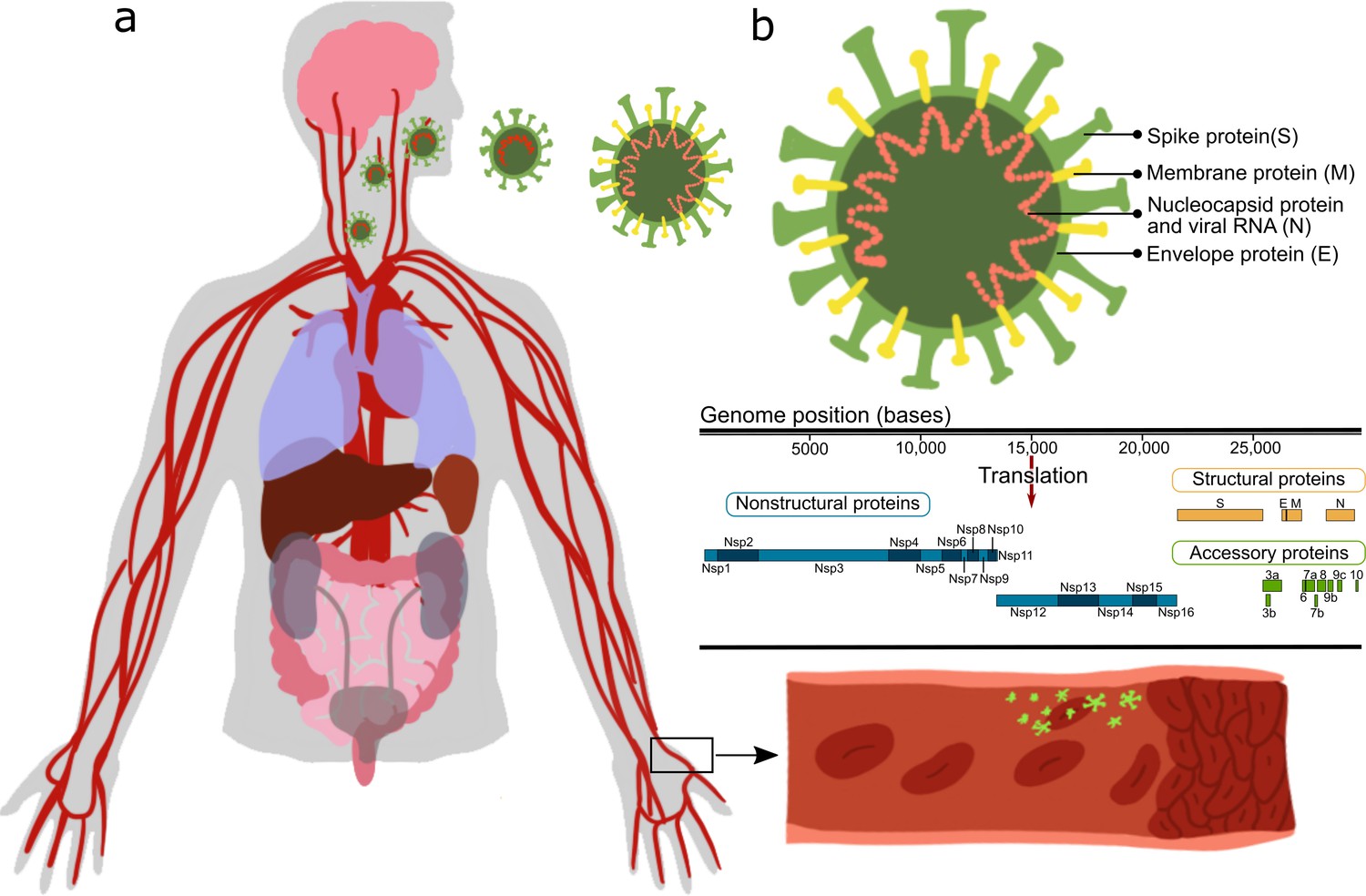
Tabella 1 – Sindrome respiratoria acuta grave (SARS)-proteine CoV-2.
| Proteine SARS-CoV-2 | Impatto generale |
|---|---|
| proteine strutturali | |
| S (picco ) | La proteina Spike, media il legame all’ACE2, la fusione con la membrana ospite La glicoproteina di superficie, deve essere elaborata dalla proteasi cellulare TMPRSS2 (Gordon et al., 2020) |
| M (membrana ) | Glicoproteina di membrana, il componente predominante dell’involucro Un importante driver per l’assemblaggio e il germogliamento del virus (Gordon et al., 2020) |
| E (envelope) | Proteina dell’involucro , coinvolta nella morfogenesi e nell’assemblaggio del virus La coespressione di M ed E è sufficiente per la formazione e il rilascio di particelle simili a virus (Gordon et al., 2020) |
| N (nucleocapside ) | La fosfoproteina nucleocapside si lega al genoma dell’RNA (Gordon et al., 2020) |
| Proteine non strutturali | |
| nsp1 | Sequenza leader, sopprime la risposta antivirale dell’ospite Antagonizza l’induzione dell’interferone per sopprimere la risposta antivirale dell’ospite (Gordon et al., 2020) |
| nsp2 | Interferisce con la segnalazione della cellula ospite, incluso il ciclo cellulare, le vie di morte cellulare e la differenziazione cellulare Può servire come adattatore per nsp3 Non essenziale per la replicazione del virus, ma la delezione di nsp2 riduce la crescita virale e la sintesi dell’RNA (Gordon et al., 2020; Procko , 2020) |
| nsp3 | Complesso nsp3–nsp4–nsp6 coinvolto nella replicazione virale Funziona come proteasi simile alla papaina (Gordon et al., 2020) |
| nsp4 | Complesso nsp3–nsp4–nsp6 coinvolto nella replicazione virale (Gordon et al., 2020) Si prevede che il complesso nuclei e ancori i complessi di replicazione virale su vescicole a doppia membrana nel citoplasma (mitocondri) |
| nsp5 | Inibisce i processi di segnalazione dell’interferone I intervenendo nel processo NF-κB e abbattendo STAT un fattore di trascrizione Funziona come proteasi simile alla 3-chimotripsina, scinde la poliproteina virale (Gordon et al., 2020) |
| nsp5_c145a | Mutante catalitico morto di nsp5 (Gordon et al., 2020) |
| nsp6 | Complesso nsp3–nsp4–nsp6 coinvolto nella replicazione virale Limita l’espansione dell’autofagosoma Componenti del complesso mitocondriale V (il complesso rigenera ATP dall’ADP) copurificano con nsp6 (Gordon et al., 2020) |
| nsp7 | Cofattore del complesso nsp12 nsp7–nsp8 in parte della RNA polimerasi (nsp7, 8, 12 – complesso di replicazione)Influenza il trasporto degli elettroni, la segnalazione GPCR e il traffico di membrana (Gordon et al., 2020; Peng et al., 2020b; Romano et al. ., 2020; Hillen et al., 2020) |
| nsp8 | Cofattore del complesso nsp12 nsp7-nsp8 in parte della RNA polimerasi. Colpisce la particella di riconoscimento del segnale e il ribosoma mitocondriale (Gordon et al., 2020; Peng et al., 2020b; Romano et al., 2020; Chi et al., 2003) |
| nsp9 | Proteina legante l’ssRNA (può legare sia il DNA che l’RNA, ma preferisce l’ssRNA) Interagisce con il complesso di replicazione (nsp7, 8, 12) (Cornillez-Ty et al., 2009) |
| nsp10 | Cofattore di nsp16 e nsp14 (Romano et al., 2020) Essenziale per l’attività della metiltransferasi nsp16 (stimolatore di nsp16) Proteina a dito di zinco essenziale per la replicazione (Gordon et al., 2020; Peng et al., 2020b) |
| nsp11 | Funzione sconosciuta |
| nsp12 | Funziona come una RNA polimerasi diretta all’RNA, la subunità catalitica colpisce lo spliceosoma (Gordon et al., 2020; Peng et al., 2020b; Romano et al., 2020; Hillen et al., 2020) |
| nsp13 | Ha attività di elicasi e 5′ trifosfatasi Avvia il primo passaggio nel capping dell’mRNA virale nsp13,14,16 installa la struttura del cap sull’mRNA virale nel citoplasma anziché nel nucleo, dove l’mRNA dell’ospite è ricoperto (Gordon et al., 2020; Peng et al., 2020b; Romano et al., 2020; Ivanov et al., 2004) |
| nsp14 | Oltre alla funzione di capping della metiltransferasi, nsp14 è anche un’endonucleasi (3’–5′ exoribonucleasi) che corregge le mutazioni durante la replicazione del genoma (Gordon et al., 2020; Peng et al., 2020b; Romano et al., 2020 ) |
| nsp15 | L’endoribonucleasi ha un’attività endonucleasica specifica dell’uridina, essenziale per la sintesi dell’RNA virale (Gordon et al., 2020; Romano et al., 2020) |
| nsp16 | Può comportare la complessazione con nsp10 e nsp14, per la stabilizzazione dell’omoenzima, per il capping dell’mRNA (Gordon et al., 2020; Peng et al., 2020b; Romano et al., 2020) |
| Cornice di lettura aperta (fattori accessori ) | |
| o rf3a | Confezionamento in virioni Media il traffico di proteine spike fornendo segnali di ritenzione ER/golgi Induce IL-6b, attiva NF-κB, attiva l’inflammasoma NLRP3 (Gordon et al., 2020) |
| o rf3b | Antagonista dell’interferone e coinvolto nella patogenesi (Gordon et al., 2020) |
| o rf6 | Antagonista dell’interferone di tipo I, sopprime l’induzione dell’interferone e le vie di segnalazione dell’interferone (Gordon et al., 2020) |
| o rf7a | Può essere correlato all’apoptosi indotta da virus (Gordon et al., 2020) |
| o rf7b | Funzione sconosciuta |
| o rf8 | Hotspot di ricombinazione Induce stress ER e attiva gli inflammasomi NLRP3 Bassa somiglianza con SAR-CoV (Gordon et al., 2020) |
| o rf9b | Sopprime la risposta antivirale dell’ospite Mira alle molecole adattatrici associate ai mitocondri MAVS e limita le risposte all’interferone della cellula ospite (Gordon et al., 2020) |
| o rf9c | Nessuna prova che questa proteina sia espressa durante l’infezione da SARS-CoV-2 (Gordon et al., 2020) |
| o rf10 | Nessuna prova che questa proteina sia espressa durante l’infezione da SARS-CoV-2 (Gordon et al., 2020) |
Per affrontare queste sfide, abbiamo coltivato cellule endoteliali della vena ombelicale umana (HUVEC) e le abbiamo trasdotte sistematicamente con particelle lentivirali che codificano 26 delle 29 proteine virali, separatamente. I tre geni rimanenti non sono stati inclusi in questo studio esclusivamente per ragioni tecniche. Abbiamo quindi esaminato i loro effetti sulla permeabilità del monostrato HUVEC e l’espressione di fattori coinvolti nella permeabilità vascolare e nella coagulazione.
I risultati sono stati analizzati nel contesto delle reti PPI virus-host e host-host. Combinando le intuizioni dei risultati sperimentali e computazionali, abbiamo generato un modello che spiega come ciascuna delle 26 proteine di SARS-CoV-2, inclusa una forma mutata di nsp5, il mutante catalitico morto chiamato nsp5_c145a, influenzi la rete proteica che regola il sistema vascolare funzionalità. Inoltre, una volta convalidato il modello PPI con i nostri dati sperimentali, lo abbiamo applicato a più di 250 proteine che sono state identificate in letteratura come colpite dalle proteine SARS-CoV-2.
Questo ci ha permesso di individuare le proteine SARS-CoV-2 più dominanti e di tracciare i loro effetti. Nel complesso, questo lavoro mostra come ciascuna delle proteine SARS-CoV-2 influisca in modo differenziale sulla funzionalità vascolare; inoltre, una volta convalidato il modello, lo abbiamo applicato per identificare come le proteine SARS-CoV-2 interagiscono con proteine che sono state significativamente correlate con i cambiamenti nella funzionalità cellulare.



{kind=link}