










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")


Patients diagnosed with a type of brain tumor survived for longer when they were treated aggressively with surgery, radiation and chemotherapy.
But far from suggesting that more treatment always leads to better survival, the study by UC San Francisco underscores the critical role of genomic profiling in diagnosing and grading brain tumors.
In the study, UCSF researchers followed 38 patients with a tumor type that was reclassified by the World Health Organization in November 2021, from a grade 2 or 3 glioma, to a “glioblastoma, IDH-wildtype, CNS WHO grade 4,” based on its molecular features.
The new more accurate diagnosis comes from genomic profiling in which the DNA alterations that drive tumor growth are identified. The previous diagnosis was determined by traditional microscopic comparisons between cancer cells and normal cells.
All patients underwent genomic sequencing using the UCSF500 Cancer Gene Panel and were offered treatment consistent with a conventional glioblastoma, the deadliest and most common adult brain tumor.
The length of their survival was compared with a retrospective cohort of 130 patients with the same tumor type, whose treatment regimens were more conservative, in line with the previous tumor classification.
The first group of patients survived an average of 24 months, while the second group survived an average of 16 months, the researchers reported in their study, appearing in the online issue of Neuro-Oncology on April 8, 2022, and publishing in the journal’s print version later this year.
“The study shows that genomic profiling resulted in more aggressive patient management that ultimately led to improved clinical outcomes, compared to the biologically matched historical patient cohort,” said senior author, David Solomon, MD, Ph.D., assistant professor in the UCSF Department of Pathology, investigator at the UCSF Brain Tumor Center, and a principal investigator of the UCSF Glioblastoma Precision Medicine Program.
Early/evolving glioblastoma may have ‘underlying biologic differences’
In studying the MRIs of the first group of patients, the researchers found that 33 of the 38 had imaging features suggestive of a lower-grade glioma, while the remaining five had imaging features suggestive of conventional glioblastoma, such as peripheral enhancement and dead tissue.
While both groups were histologically and molecularly indistinguishable, the first set, so-called early/evolving glioblastoma, may have “underlying biologic differences” in the immune micro-environment and an intact blood-brain barrier that may affect treatment efficacy.
The UCSF research was prompted by a landmark 2015 study by the Cancer Genome Atlas Research Network that performed genome-wide analyses on 293 grade 2 and 3 gliomas. The researchers identified a subset of patients, approximately one in five, who lacked an IDH mutation, a molecular biomarker known to be associated with better outcomes.
“While these patients did not have the traditional histological hallmarks of conventional glioblastoma, they shared the same molecular features and had similar survival,” said Solomon, who has pioneered genomic profiling for diagnostic classification and treatment of brain tumors at UCSF since 2015, and has co-authored more than 50 publications on brain tumor molecular pathogenesis.
Study solves survival mystery for doctors treating lower-grade glioma
The results of this study not only led to the treatment objective of the UCSF study, but it also solved a conundrum that had bedeviled neuro-oncologists for years: why some grade 2 and 3 glioma patients progressed slowly and survived for several years, while others advanced rapidly and died within a year or two.
“Historically, we’ve relied on what pathologists see under the microscope to guide treatment, which can be subjective to the pathologist’s experience and the size of the sample collected from the neurosurgeon, said co-author Jennie Taylor, MD, MPH, a neuro-oncologist with the UCSF Department of Neurological Surgery who treats adult brain tumor patients.
Future research may further extend the survival of patients with IDH-wildtype glioblastoma, said Taylor, who is also affiliated with the UCSF Weill Institute for Neurosciences.
“Integrating genomic profiling into the pathology report – which takes a small quantity of brain tumor cells—means that doctors can be more confident in their treatment recommendations, enabling more patients to take part in clinical trials.”
When the UCSF500 Cancer Gene Panel was first implemented in 2015, around 5 to 10% of patients had to have their pathological diagnosis amended following genomic testing, Solomon said. In pediatrics, the figure was even higher, affecting six of the first 31 patients who were tested, according to a 2016 study. Today, genomic sequencing is routinely carried out for all adult and pediatric brain tumors at UCSF to ensure accurate diagnosis and optimal care. It is usually covered by both private and public insurers.
A preliminary pathological diagnosis following microscopic review is made after tumor resection or biopsy. A final diagnosis integrating the molecular findings comes about three weeks later, but radiation or chemotherapy may be initiated earlier if recommended.
Sometimes the final diagnosis is sobering, and sometimes it is joyful – such as the child whose earlier microscopic evaluation had pointed toward a grade 4 tumor, which was corrected to grade 1 tumor after genomic testing, said Solomon.
“There’s no question that we need a definitive diagnosis to provide the most appropriate treatment plan. That might mean more treatment and it might mean less.”
Gliomas comprise a very heterogeneous group of primary tumors of the central nervous system (CNS), originally classified according to their histological similarity or the supposed origin of non-neoplastic glial cells (1). The common presentation of gliomas includes seizures, cranial nerve palsies, visual field defects, language dysfunction, or increased intracranial pressure. Gliomas are considered to originate from glial (progenitor) or stem cells that develop glial characteristics. Most gliomas are characterized by diffuse infiltrative growth of tumor cells in the preexisting parenchyma of the CNS (2). Gliomas are traditionally divided into two main categories: Diffuse gliomas (DGs) and non-diffuse gliomas (NDGs) (3).
DGs are one of the most frequent primary neoplasms (4). They represent approximately 81% of all malignant brain tumors and have a high mortality and morbidity rate (5). In these tumors, the degree of malignancy is assigned based on pathological features (6). The main classes of DGs are astrocytomas, oligodendrogliomas, oligoastrocytomas and glioblastoma multiforme, and are classified according to the criteria of the World Health Organization (WHO) as grades II, III or IV (6,7).
In 2016, WHO updated its classification and incorporated the detection of distinctive molecular alterations to traditional histological methods (8). The use of ‘integrated’ phenotypic and genotypic parameters for classification added greater objectivity in the diagnostic process (9). This has allowed defining more biologically homogeneous diagnostic entities, leading to better patient management and more precise indications of response to treatment.
The 2016 WHO classification of DGs has incorporated two molecular biomarkers: The mutation status of the isocitrate dehydrogenase (IDH) gene (the IDH1 (codon 132) and IDH2 (codon 172) genes) and the presence or absence of the 1p/19q co-deletion (complete deletion of both the short arm of chromosome 1 and the long arm of chromosome 19) (10).
When these molecular biomarkers could not be performed, the term NOS (not otherwise specified) should be added to indicate that the study has been inconclusive (8,11). The new glioma classification allows differentiating astrocytomas and glioblastomas according to the IDH1 or IDH2 mutation status and allows diagnosing tumors with 1p/19q co-deletion and IDH1 or IDH2 mutation as oligodendrogliomas (Fig. 1).
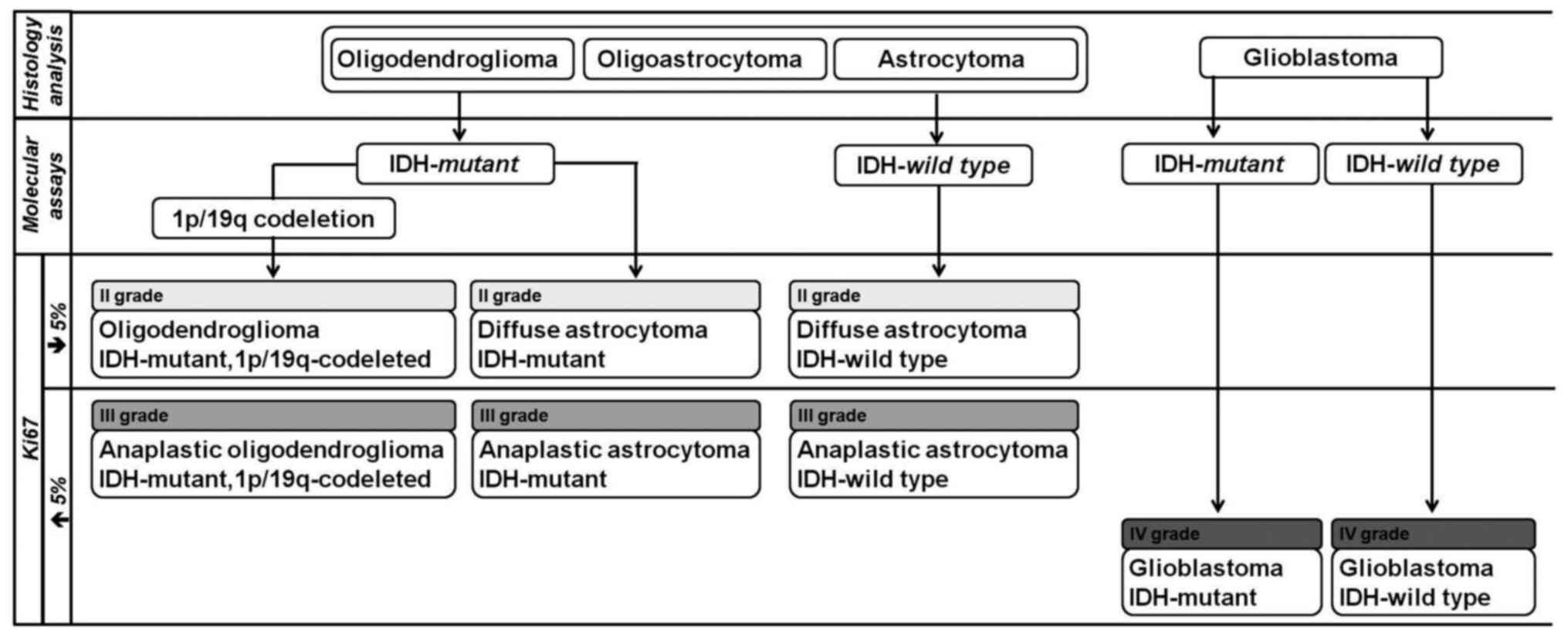
Simplified algorithm for integrated diagnosis of diffuse gliomas (2016 World Health Organization classification). IDH, isocitrate dehydrogenase.
The IDH status (mutant or wild type) is very relevant for the diagnosis of gliomas. The most common variant (~88%) is a substitution of the amino acid Arginine to Histidine in codon 132 of the IDH1 gene (IDH1-R132H) (12). Less common variants such as R132C, R132M, and R132K, as well as a substitution in the IDH2 gene (R172H, R172C, R172M and others), have also been described (13). The IDH status allows identifying a subgroup of glial tumors that have a better survival prognosis (14).
The presence of the 1p/19q co-deletion is associated with greater survival and better response to chemotherapy (15). This co-deletion is observed mainly in oligodendrogliomas, and is useful to minimize the diagnosis of oligoastrocytomas, which are tumors exhibiting mixed histology (16). The WHO classification discourages this diagnosis as molecular genetic analyses have proved to be efficient to re-classify them.
In this study, we analyzed and evaluated This study aimed to evaluate the impact caused by the new diagnostic classification of gliomas in a series of cases studied in Argentina. In addition, we considered the possible associations between erroneous tumor classification, histological characteristics and survival data.
referencelink : https://www.spandidos-publications.com/10.3892/mco.2021.2312
More information: Yalan Zhang et al, Prospective genomically-guided identification of ‘early/evolving’ and ‘undersampled’ IDH-wildtype glioblastoma leads to improved clinical outcomes, Neuro-Oncology (2022). DOI: 10.1093/neuonc/noac089



{kind=link}