










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



A Japanese research team has developed a cyclic peptide (a chain of amino acids bonded circularly) that enhances blood-brain barrier (BBB) penetration.
By attaching the cyclic peptide to the surface of nanoparticles, research and development of new drug nanocarriers for drug delivery to the brain becomes possible.
Unlike blood circulation to the peripheral organs in the body, the BBB prevents various substances, including many drugs, from moving from the blood into the brain.
Biopharmaceuticals and macromolecular drugs are attracting attention as new treatments for previously untreatable diseases and for improving outcomes.
However, these high molecular weight drugs are unable to penetrate the BBB.
Technologies that can deliver them to the brain would bring significant progress in the development of medications that act on the brain.
Aiming to develop technologies applicable to various drugs, a research team from Kumamoto University, Japan worked on developing a cyclic peptide able to penetrate the BBB.
In their search to find a peptide with the desired function, they turned to viruses called phages. From a phage library listing cyclic peptides with 109 types of amino acid sequences, the researchers searched for phages able to penetrate human BBB model cells and analyzed their sequences.
Since the size of a phage (about 1,000 nanometers) is larger than macromolecular drugs, the scientists expected that these cyclic peptides would also allow drug penetration into the BBB.

Of the two new cyclic peptides they discovered, one promoted phage penetration not only in human BBB model cells but also in monkey and rat BBB model cells.
Furthermore, this phage could be found in the brain of a mouse 60 minutes after intravenous injection. In additional experiments, the researchers modified liposomes by adding the cyclic peptide to the surface of liposomes thereby creating 150 nanometer-sized artificial nanoparticles.
When this modified liposome was injected intravenously into a mouse, it was also detected in the brain 60 minutes later showing that the new cyclic peptide facilitates penetration of phage and liposome nanoparticles through the BBB allowing for delivery into the brain.
“Liposomes are nanocarrier that can encapsulate various substances. The liposome whose surface has been modified with this new cyclic peptide can be used as a nanocarrier to bypass the BBB. A way to deliver macromolecular drugs to the brain has been opened,” said Professor Ohtsuki. “We expect this research to contribute significantly toward the development of drugs for central nervous system diseases, including Alzheimer’s disease.”
Progress in the development of new pharmaceuticals for the treatment of the brain in aging, including Alzheimer’s disease (AD), has been slow because the products of biotechnology, recombinant proteins or gene therapies, are all large molecule pharmaceuticals that do not cross the blood-brain barrier (BBB). In 2019, there is not a single recombinant protein that is FDA approved for the treatment of AD, or any other brain disease, wherein the drug must cross the BBB to enter the brain.
Bevacizumab (Avastin®) is FDA approved for brain cancer (Han et al., 2014), but this monoclonal antibody (MAb) does not cross the non-disrupted BBB (Liu et al., 2016), and works by sequestration of vascular endothelial growth factor (VEGF) within the blood volume of the tumor. Natalizumab (Tysabri®) is FDA approved for the treatment of multiple sclerosis (Cadavid et al., 2013), but this MAb does not cross the BBB and works by blocking the trafficking of lymphocytes across the brain endothelial wall (Engelhardt and Coisne, 2011).
In 2019, an adeno-associated virus (AAV)-9-based gene therapy was approved for the treatment of infantile spinal muscular atrophy (SMA)-1 with a 1-time intravenous (IV) administration of the self-complementary (sc)-AAV9 encoding the survival motor neuron type 1 (SMN1) gene (Mendell et al., 2017).
This is the first FDA approved biologic for a brain disease that crosses the BBB. The development of this new treatment for SMA did not arise from a rational drug delivery platform, but rather was a product of the serendipitous finding that the AAV9 serotype traverses the BBB (Foust et al., 2009). However, as discussed below, there are limitations to the delivery of therapeutic genes to the brain with the AAV platform.
The discovery that certain AAV serotypes, such as AAV9, undergo transport across the BBB is a model of past efforts in the discovery of BBB-penetrating brain pharmaceuticals. That is, transport across the BBB was discovered serendipitously, rather than being an outgrowth of a focused effort in BBB transport technology.
L-DOPA was developed as a treatment for Parkinson’s disease (PD) after the incidental finding that this amino acid precursor of dopamine, the neurotransmitter deficient in PD, penetrated the BBB following systemic administration (Davidson et al., 1971). L-DOPA is a large neutral amino acid and the mechanism of L-DOPA transport across the BBB was subsequently shown to be carrier-mediated transport via the BBB large neutral amino acid transporter (Wade and Katzman, 1975), and L-DOPA transport is mediated via the large neutral amino acid transporter type 1 (LAT1; Kageyama et al., 2000).
The LAT1 transporter is selectively expressed at the BBB (Boado et al., 1999). Similarly, gabapentin, a cyclic gamma-amino acid, was discovered to penetrate the brain (Vollmer et al., 1986), and this was later shown to be mediated by the LAT1 transporter at the BBB (Dickens et al., 2013).
The brain drug development of brain-penetrating small molecules, recombinant proteins, or gene medicines is generally not successful unless the pharmaceutical crosses the BBB. Brain drug development should be executed as a binary process with parallel advancement in both brain drug discovery and BBB drug delivery, but this has not been the practice of the pharmaceutical industry. What has taken place is a primary focus on brain drug discovery with a minimal, if any, parallel effort in BBB drug delivery.
As a consequence, there are few present-day FDA approved biologics for the serious diseases of the brain in aging, such as AD or PD. This situation is not expected to change for at least a generation if current practices continue and brain drug development operates in a BBB-free zone.
This review will outline present-day approaches to brain drug delivery and discuss a platform for future brain drug development that merges BBB drug delivery technology with brain drug and gene discovery.
Biologic Treatments of Alzheimer’s Disease Illustrate the Need for BBB Drug Delivery
The dementia of AD is caused by the deposition of Abeta amyloid plaque in the brain (Cummings and Cotman, 1995; Näslund et al., 2000), which arises from the abnormal processing of the amyloid precursor protein (APP).
The amyloid hypothesis emerged in 1984, following the isolation and N-terminal amino acid sequencing of the 43 amino acid Abeta amyloid peptide, which has a single carboxyl-terminal threonine residue (Glenner and Wong, 1984). In the intervening 35 years, there is not a single FDA approved therapeutic for AD that reverses the dementia of AD secondary to intervention in the formation of the Abeta plaque.
Clinical trials of drugs, such as anti-amyloid antibodies (AAA), aimed at the reduction of amyloid plaque in the brain in AD have not led to FDA approval. This dismal record in AD drug development has led many to question the validity of the amyloid hypothesis of AD (Panza et al., 2019).
However, the failure of these clinical trials should not be used to refute the validity of the amyloid hypothesis of AD, if the anti-amyloid drug never reached the intended target owing to lack of BBB transport.
If a drug aimed at brain amyloid, or any other target in the brain, does not cross the BBB, then no success in the clinical trial can be expected, and the amyloid hypothesis of AD has not been tested by the clinical trial. Multiple AAAs have failed in AD clinical trials because in all cases, the drugs do not cross the BBB, and no BBB drug delivery technology was introduced in AAA drug development (Pardridge, 2019).
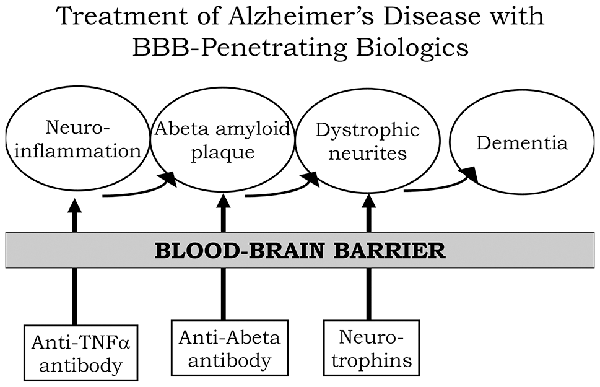
A process by which amyloid plaque accumulation in the brain in AD leads to dementia is depicted in Figure 1. Dementia and cognitive decline of AD is not caused by amyloid plaque, per se, because amyloid plaque accumulation occurs before the onset of cognitive decline; the amyloid plaque leads to neurite dystrophy and synaptic loss, which then leads to dementia (Serrano-Pozo et al., 2011).
If plaque reduction is not followed by repair of dystrophic neurites, then no improvement in dementia can be expected. AD is a chronic neuro-inflammatory condition mediated by proinflammatory cytokines, such as tumor necrosis factor (TNF)-α (McAlpine and Tansey, 2008). Therefore, a reversal of plaque formation may be followed by the further accumulation of plaque if the underlying neuroinflammation in the brain is not treated (Figure 1). Biologic drugs are currently available that can intervene in all three steps causing the dementia of AD (Figure 1).
Neuro-inflammatory cytokines, such as TNFα, can be blocked by delivery to the brain of biologic TNF inhibitors (TNFI), such as anti-TNFα antibodies (Humira®, Remicade®), or TNFα decoy receptors (Enbrel®). However, these biologic TNFIs do not cross the BBB (Pardridge, 2010), and industry has made little attempt to re-engineer these agents for BBB delivery. The amyloid plaque of AD can be reduced by AAAs following direct intracerebral injection of the antibody (Solomon et al., 1997).
In these early studies, the AAA was injected directly into the brain, because the AAA does not cross the BBB. However, the clinical trials for AAAs, e.g., bapineuzumab, solanezumab, gantenerumab, aducanumab, and others, administered the AAA by IV infusion on the assumption that the AAA crossed the BBB. When BBB transport of AAAs is measured, no BBB transport is observed in the absence of BBB delivery technology (Boado et al., 2007).
The AAAs entered AD clinical trials with IV infusion, because industry made no attempt to develop BBB delivery technology to enable the re-engineering of these AAAs for BBB transport. With respect to repair of dystrophic neurites, neurotrophins, such as brain-derived neurotrophic factor (BDNF), ciliary neurotrophic factor (CNTF), fibroblast growth factor (FGF)-2, and others, can enhance neuronal repair in neurodegeneration (Kazim and Iqbal, 2016).
However, the BDNF, CNTF, or FGF2 clinical trials for brain disease failed, because these agents do not cross the BBB (Pardridge, 2015a). What is needed for the treatment of the dementia of AD is a combination drug therapy that simultaneously aims to reduce neuroinflammation in the brain, disaggregate amyloid plaque, and repair dystrophic neurites.
However, in the case of all three classes of biologic drugs for the brain, the neurotherapeutic needs to be re-engineered for BBB drug delivery (Figure 1). Such re-engineering platforms are available as discussed below in the context of molecular Trojan horses for BBB drug delivery of biologics. Prior to the discussion of transvascular drug delivery to the brain, it is necessary to review intra-thecal drug delivery to the brain, since this has been the default approach to brain drug delivery for decades.
Brain Drug Delivery Into Cerebrospinal Fluid
Bulk Flow of Cerebrospinal Fluid Within the Brain
The surface of the human brain is bathed with about 140 ml of cerebrospinal fluid (CSF). The CSF is secreted by the choroid plexus at each of the four ventricles of the brain (two lateral ventricles (LVs), a 3rd ventricle, and a 4th ventricle), and the entire volume of CSF is produced every 4–5 h or 4–5 times per day in the human brain (Pardridge, 2016).
This CSF is rapidly exported to the blood via absorption into the superior sagittal sinus across the arachnoid villi. Owing to this rapid egress of CSF from brain to blood, intrathecal injection of the drug into CSF is similar to a slow IV infusion (Fishman and Christy, 1965). In contrast to the rapid rate of bulk flow of CSF out of the cranium, the diffusion of the drug into brain parenchyma from the CSF is limited, because diffusion decreases with the square of the diffusion distance. Consequently, drug is distributed only to the ependymal surface of brain following injection into CSF, and not into the brain parenchyma (Pardridge, 2016).
The intra-thecal route of drug delivery to brain is suitable for treatment of diseases that affect the surface of the brain, such as carcinomatous meningitis, but is not able to deliver drug into the parenchyma of brain without exposing the surface of the brain to high, and generally toxicologic, drug concentrations (Yamada et al., 1991; Day-Lollini et al., 1997).
Different Routes of Drug Injection Into CSF: Lumbar, Ventricular, or Cisternal
The simplest route of drug injection into CSF is a lumbar puncture. However, MRI studies in the primate show this route of delivery may treat the surface of the spinal cord, but very little drug reaches the cerebral hemispheres (Ohno et al., 2019).
An alternative route of CSF injection is intra-cerebroventricular (ICV) delivery following the implantation of an Ommaya reservoir into one of the two LVs (Ommaya, 1963). Recently FDA approval was granted for the treatment of Batten disease type 2, a childhood lysosomal storage disorder caused by mutations in the gene encoding the lysosomal enzyme, tripeptidyl peptidase (TPP)-1, by ICV delivery using an Ommaya reservoir (Schulz et al., 2018).
Recombinant human TPP1 (Brineura®) is infused into one LV every 2 weeks. Approval was granted because of improvement in peripheral motor function, although no effect of drug treatment on dementia, seizures, or blindness of Batten disease type 2 was recorded. The TPP1 enzyme is taken up by peripheral tissues via the mannose 6-phosphate receptor (M6PR), and delivery of TPP1 to peripheral tissues following injection into an Ommaya reservoir would be expected as the enzyme passes rapidly from CSF to peripheral blood, and then to the M6PR of peripheral tissues.
A peripheral mechanism of action cannot be excluded, because there was no control arm in the clinical trial that administered the TPP1 enzyme by IV infusion (Schulz et al., 2018). Apart from rapid drug delivery to the peripheral bloodstream, other limitations of the Ommaya reservoir include the poor distribution of the drug to the contralateral side of the brain, owing to minimal retrograde flux of CSF from the 3rd ventricle to the opposite LV (discussed below).
The third route of CSF injection that achieves the best distribution of drugs to both the bilateral forebrain and the spinal cord is a cisternal injection in the cerebro-medullary cistern (CMC; Ohno et al., 2019). However, this route of injection is technically difficult, is near vital structures of the brain, and has yet to enter into clinical practice. Still, a CMC injection would be expected to deliver the drug into brain parenchyma by reliance on drug diffusion from the CSF surface of the brain.
Diffusion as the Primary Mechanism for Drug Penetration Into the Brain From CSF
Following drug injection into the CSF compartment, the drug may gain access to brain parenchyma via one of two mechanisms:
(a) diffusion; or
(b) bulk flow through perivascular spaces (Pardridge, 2016).
If diffusion is the primary mechanism, then a logarithmic decline in drug concentration would be expected as drug diffuses into the brain from the CSF compartment; this is because diffusion decreases with the square of the diffusion distance. Conversely, if bulk flow through perivascular spaces was the predominant pathway, then a more uniform distribution of the drug in the brain would be observed with minimal decline in drug concentration within the parenchyma as compared to the drug concentration in CSF.
The rate of bulk flow through perivascular space is low, 0.2 μl/min, and only about 5% the rate of CSF flow in the rat, 3.4 μl/min (Pardridge, 2016). The evidence accrued over the last 40 years of research on CSF drug delivery to brain indicates diffusion is the primary mechanism for drug movement into the brain from the CSF surface. Blasberg et al. (1975) injected small molecule drugs into one LV of the primate, and then measured the drug concentration at each mm of brain removed from the CSF surface.
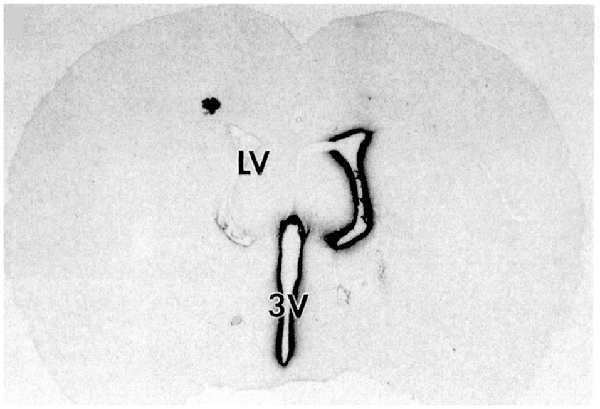
A steep logarithmic decline in drug concentration in brain parenchyma was observed. The concentration in the brain of thiotepa, a small molecule, was only 1% of the CSF concentration at just 1 mm of distance removed from the CSF surface. The poor distribution of drugs into brain parenchyma following injection into a LV is illustrated with brain autoradiography (Figure 2).
The distribution of BDNF in rat brain was measured with autoradiography of brain obtained 20 h after the ICV injection of [125I]-BDNF in the LV (Yan et al., 1994). As shown in Figure 2, the BDNF has diffused only 0.2 mm into the parenchyma ipsilateral to the LV injection, with no measurable BDNF in the contralateral brain. This is because after drug is injected into an LV, the drug moves by CSF bulk flow to the third ventricle (3V), with minimal reflux up into the contralateral LV, then moves to the 4th ventricle, then over the surface of the brain, where it is absorbed into the blood.
As noted by Fishman and Christy (1965) over 50 years ago, intrathecal injection of the drug is equivalent to an IV infusion. An autoradiographic result similar to that shown in Figure 2 was demonstrated following the LV injection of [125I]-insulin-like growth factor-1 (IGF1) in the rat (Nagaraja et al., 2005).
A detailed study of monoclonal antibody (MAb) distribution in the brain following ICV administration was reported for the cynomolgus monkey following continuous 24/7 ICV infusion of the MAb in one LV for 42 consecutive days (Yadav et al., 2017). The MAb targeted the beta secretase-1 (BACE1) and was developed as a therapy for AD. The study made several findings:
• The maximal MAb concentration, Cmax, in plasma was 3 μM following ICV infusion and 12 μM following IV infusion. However, the infusion dose (ID) was about 4-fold higher over the course of the study for the IV route as compared to the ICV route. Therefore, a comparable distribution of MAb in plasma was obtained with either the ICV or IV route, thus confirming the observations of Fishman and Christy (1965) that drug injection into the CSF is equivalent to an IV administration.
• The concentration of the MAb in the contralateral motor cortex, which is near the CSF surface, was nearly 30-fold higher than the MAb concentration in a deep parenchyma structure, the contralateral putamen. If the perivascular bulk flow was prominent, there should be little difference in MAb concentration between cortical and subcortical regions of the brain.
• Given a diffusion coefficient of 0.7 × 10−6 cm2/s, for a large molecule such as a MAb (Pardridge, 2016), the effective diffusion distance over a 6 week period would have a radius of 16 mm and a diameter of 32 mm, and these distances are comparable to the diameter, 40 mm, of a monkey brain (Pardridge, 2016). Thus, continuous 24/7 ICV infusion for 42 days would enable diffusion alone to cover most of the primate brain.
• The distribution of the MAb in the brain was detected with immunocytochemistry, which showed the MAb did not penetrate the white matter of the brain (Yadav et al., 2017). However, perivascular flow in the brain occurs in white matter (Hladky and Barrand, 2014). If the perivascular flow was a prominent mechanism for MAb distribution into brain tissue from the CSF, then MAb should have penetrated into the white matter of the brain.
The minimal distribution of a biologic into human brain following an ICV injection was demonstrated in humans with whole-body positron emission tomography (PET). A [124I]-labeled 8H9 MAb was injected into the LV with an Ommaya reservoir of a patient with neuroblastoma metastatic to the meninges (Larson et al., 2015).
Whole-body PET scanning at 24 h after the ICV injection showed MAb sequestration by meningeal cancer on the surface of the brain or spinal cord, but no MAb penetration into brain parenchyma. MAb was readily detected in the liver at 24 h following the ICV injection.
Uptake of the MAb by the liver following the ICV injection is expected since the injection of a drug into CSF is equivalent to an IV infusion of the drug (Fishman and Christy, 1965).
Source:
Kumamoto University



{kind=link}