










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Le prove di un ampio studio su diverse migliaia di pazienti mostrano che gli uomini hanno concentrazioni più elevate di enzima 2 (ACE2) che converte l’angiotensina nel sangue rispetto alle donne.
Poiché ACE2 consente al coronavirus di infettare le cellule sane, questo può aiutare a spiegare perché gli uomini sono più vulnerabili al COVID-19 rispetto alle donne.
Lo studio, pubblicato oggi sull’European Heart Journal , ha anche scoperto che i pazienti con insufficienza cardiaca che assumevano farmaci mirati al sistema renina-angiotensina-aldosterone (RAAS), come gli inibitori dell’enzima di conversione dell’angiotensina (ACE) o i bloccanti del recettore dell’angiotensina (ARB) non hanno concentrazioni più elevate di ACE2 nel sangue.
Adriaan Voors (MD-Ph.D.), Professore di cardiologia presso il centro medico universitario di Groningen (Paesi Bassi), che ha guidato lo studio, ha dichiarato:
“I nostri risultati non supportano l’interruzione di questi farmaci nei pazienti COVID-19 come è stato suggerito da precedenti rapporti.”
Alcune ricerche recenti hanno suggerito che gli inibitori di RAAS potrebbero aumentare le concentrazioni di ACE2 nel plasma – la parte liquida del sangue – aumentando così il rischio di COVID-19 per i pazienti cardiovascolari che assumono questi farmaci.
L’attuale studio indica che non è così, sebbene abbia esaminato solo le concentrazioni di ACE2 nel plasma, non in tessuti come il tessuto polmonare. Inoltre, lo studio non può fornire prove definitive sugli effetti degli inibitori di RAAS in pazienti con COVID-19.
Le sue conclusioni sono principalmente limitate ai pazienti con insufficienza cardiaca e i pazienti non avevano COVID-19, quindi i ricercatori non possono fornire un collegamento diretto tra il decorso della malattia e le concentrazioni plasmatiche di ACE2.
Il prof Voors ha dichiarato: “L’ACE2 è un recettore sulla superficie delle cellule. Si lega al coronavirus e gli consente di entrare e infettare le cellule sane dopo che è stato modificato da un’altra proteina sulla superficie della cellula, chiamata TMPRSS2.
Alti livelli di ACE2 sono presenti nei polmoni e, pertanto, si ritiene che svolgano un ruolo cruciale nella progressione dei disturbi polmonari correlati a COVID-19 “.
Il prof. Voors e i suoi colleghi stavano già studiando le differenze nei marcatori di malattia nel sangue tra uomini e donne prima dell’epidemia di coronavirus. I risultati sono diventati disponibili subito dopo l’inizio della pandemia.
La prima autrice dello studio, la dott.ssa Iziah Sama dell’UMC Groningen, ha dichiarato: “Quando abbiamo scoperto che uno dei biomarcatori più potenti, l’ACE2, era molto più alto negli uomini che nelle donne, mi sono reso conto che questo aveva il potenziale per spiegare perché per gli uomini era più probabile che venissero contagiati da COVID-19 rispetto alle donne. ”
I ricercatori hanno misurato le concentrazioni di ACE2 nei campioni di sangue prelevati da due gruppi di pazienti con insufficienza cardiaca provenienti da 11 paesi europei.
Nel primo gruppo c’erano 1485 uomini e 537 donne, la coorte indice, progettata per testare le ipotesi dei ricercatori e le domande di ricerca. Quindi i ricercatori hanno convalidato i loro risultati in un secondo gruppo di 1123 uomini e 575 donne, la coorte di validazione.
L’età media (media) dei partecipanti alla coorte indice era di 69 anni per gli uomini e 75 anni per le donne, e nella coorte di validazione era rispettivamente di 74 e 76 anni.
Quando i ricercatori hanno esaminato una serie di fattori clinici che potrebbero svolgere un ruolo nelle concentrazioni di ACE2, incluso l’uso di ACE-inibitori, ARB e antagonisti del recettore dei mineralcorticoidi (MRA), nonché una storia di malattia polmonare ostruttiva cronica, arteria coronarica by- dopo l’innesto e la fibrillazione atriale, hanno scoperto che il sesso maschile era il predittore più forte di elevate concentrazioni di ACE2.
Nella coorte dell’indice, gli ACE-inibitori, gli ARBS e gli MRA non erano associati a maggiori concentrazioni plasmatiche di ACE2, e nella coorte di validazione, gli ACE-inibitori e gli ARB erano associati a basse concentrazioni di ACE2, mentre gli MRA erano associati debolmente solo a concentrazioni più elevate.
“Per quanto ne sappiamo, questo è il primo studio sostanziale che esamina l’associazione tra le concentrazioni plasmatiche di ACE2 e l’uso di bloccanti del sistema renina-angiotensina-aldosterone in pazienti con malattie cardiovascolari.
Non abbiamo trovato prove che gli ACE-inibitori e gli ARB fossero collegati ad aumentate concentrazioni di ACE2 nel plasma.
In effetti, hanno previsto concentrazioni più basse di ACE2 nella coorte di validazione, sebbene non l’abbiamo visto nella coorte dell’indice ”, ha affermato il prof. Voors.
“L’effetto degli MRA sulle concentrazioni di ACE2 non è chiaro, poiché il debole aumento delle concentrazioni nella coorte di validazione non è stato visto nella coorte dell’indice. I nostri risultati non suggeriscono che gli MRA debbano essere sospesi nei pazienti con insufficienza cardiaca che sviluppano COVID-19. Sono un trattamento molto efficace per l’insufficienza cardiaca e gli effetti ipotetici sull’infezione virale devono essere attentamente valutati rispetto ai loro comprovati benefici “, ha detto.
L’ACE2 si trova non solo nei polmoni, ma anche nel cuore, nei reni e nei tessuti che rivestono i vasi sanguigni e ci sono livelli particolarmente elevati nei testicoli. I ricercatori ipotizzano che la sua regolamentazione nei testicoli potrebbe in parte spiegare maggiori concentrazioni di ACE2 negli uomini e perché gli uomini sono più vulnerabili a COVID-19.
Altre limitazioni dello studio includono il fatto che i ricercatori hanno misurato solo le concentrazioni di ACE2 nel plasma, non nei tessuti, quindi non possono essere sicuri che le concentrazioni nel sangue siano simili a quelle osservate nei tessuti; è l’ACE2 nei tessuti polmonari che si ritiene sia importante per l’infezione virale dei polmoni, non le concentrazioni di ACE2 nel sangue.
In un editoriale di accompagnamento, il professor Gavin Oudit, dell’Università di Alberta, in Canada, e il professor Marc Pfeffer, del Brigham and Women’s Hospital, Harvard Medical School, USA, scrivono:
“Di fronte alla rapida pandemia COVID-19 in rapida espansione e in assenza di dati definitivi, i risultati di Sama et al ottenuti nei pazienti con insufficienza cardiaca nel periodo pre-COVID-19 offrono prove a sostegno del proseguimento di ACE-inibitori o ARB nei pazienti a rischio di infezione da SARS-CoV-2. Tuttavia, questo campo si sta muovendo così rapidamente che ora abbiamo due studi osservazionali sull’uso di inibitori ARB / ACE in pazienti COVID-19 ospedalizzati che non mostrano alcun rischio aumentato per i pazienti COVID-19 e che suggeriscono anche possibili benefici. “
Lo studio è uno dei numerosi articoli di ricerca, recensioni cliniche, editoriali e articoli di discussione su COVID-19 e le malattie cardiovascolari che saranno pubblicati in un numero speciale dell’European Heart Journal giovedì 14 maggio.
Secondo il Center for Systems Science and Engineering presso la Johns Hopkins University, a partire dal 12 aprile 2020, la pandemia di sindrome respiratoria acuta grave Coronavirus-2 (SARS-CoV-2) ha causato 108.867 decessi in tutto il mondo, per un totale di 1.777.666 persone infette (https://coronavirus.jhu.edu/map.html). Solo per fare un confronto, il virus SARS-Cov negli anni 2002-2003 ha infestato circa 8.500 persone in 27 paesi e causato 866 morti. [1]
Il tremendo impatto dell’infezione da SARS-Cov-2 e la scarsità o la mancanza di misure terapeutiche consolidate sta generando studi di base e clinici per esplorare i meccanismi di ingresso virale nel corpo umano e le conseguenti implicazioni patofisiologiche e terapeutiche.
La presente recensione discute il ruolo dei recettori dell’enzima 2 (ACE2) di conversione dell’angiotensina che non sono solo la porta attraverso la quale il virus entra nelle cellule, [2,3] ma anche il conduttore di diverse reazioni fisiopatologiche associate alle caratteristiche cliniche della malattia , con potenziali implicazioni terapeutiche.
Ingresso di SARS-CoV-2 nelle cellule
L’ingresso di SARS-CoV-2 nelle cellule è mediato dall’efficace legame della proteina virale spike (S), una proteina lunga da 1273 aminoacidi che appartiene all’involucro virale e sporge verso l’esterno con un aspetto simile a una “corona”, al recettori dell’enzima 2 di conversione dell’angiotensina (ACE2). [2,3]
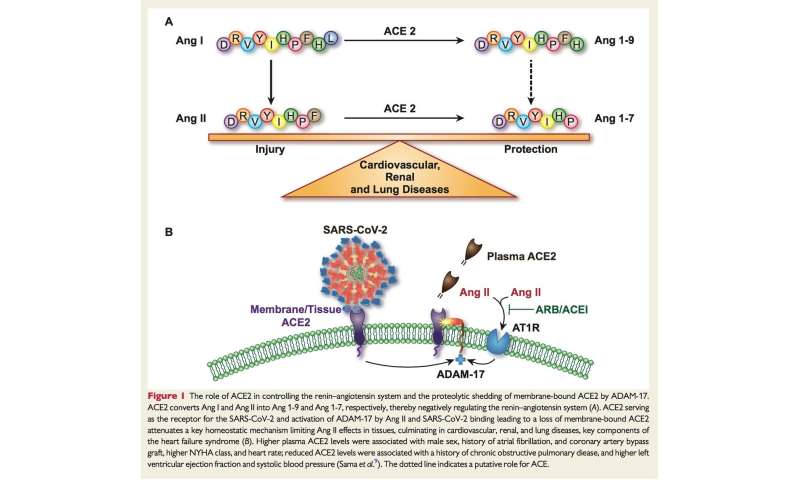
Il recettore ACE2, scoperto da due gruppi indipendenti nel 2000, [4,5] è una glicoproteina di tipo I trans-membrana (monocarbossipeptidasi) composta da 805 aminoacidi che utilizza un singolo dominio catalitico extracellulare per rimuovere un singolo aminoacido da l’ottapeptide angiotensina II per generare angiotensina1-7.
Il recettore ACE2 converte anche l’angiotensina I in angiotensina1-9, che a sua volta viene convertita in angiotensina1-7 da ACE e neprilisina (Figura 1). L’efficienza catalitica dell’ACE2 è 400 volte più alta sull’angiotensina II rispetto all’angiotensina I. [6] L’ACE2 mostra un’identità strutturale del 40% rispetto all’ACE, [4] sebbene gli ACE-inibitori non blocchino l’ACE2 a causa della diversa struttura conformazionale del catalizzatore luogo. [4]

ACE2 media l’ingresso cellulare di tre ceppi di coronavirus: SARS-CoV, NL63 e SARS-CoV-2. [7] In particolare, SARS-CoV e Sars-CoV2 condividono un’identità del 76% nella sequenza degli amminoacidi, [8] spiegando così la propensione di questi virus al legame con ACE2.
Sono state identificate alcune variazioni strutturali di ACE2 umano caratterizzate da una minore affinità di legame con la proteina virale di picco, con potenziali implicazioni protettive. [9]
Il primo passo del processo di ingresso virale è il legame della porzione N-terminale dell’unità proteica virale S1 a una tasca del recettore ACE2. Il secondo passo, che si ritiene sia della massima importanza per l’ingresso virale, è la scissione proteica tra le unità S1 e S2, che è gestita dal recettore transmembrana proteasi serina 2 (TMPRSS2), un membro della sottofamiglia Hepsin / TMPRSS. [10]
TMPRSS2 è stechiometricamente contiguo al recettore ACE2. [10] La scissione della proteina virale da parte di TMPRSS2 è un passaggio cruciale perché, dopo il distacco di S1, la rimanente unità S2 virale subisce un riarrangiamento conformazionale che guida e completa la fusione tra la membrana virale e cellulare, con successivo ingresso del virus nella cellula , rilascio del suo contenuto, replicazione e infezione di altre cellule.
L’importanza di TMPSRR2 è supportata dall’evidenza che l’ingresso di SARS-CoV e SARS-CoV-2 nelle cellule è parzialmente bloccato da mesostatato di camostat, un inibitore di TMPSRR2. [2]
Sito dei recettori ACE2
I geni ACE2 si associano al cromosoma X, [11] la sua espressione sembra essere più alta in Asia che in bianchi e afro-americani, [12] e i recettori sono onnipresenti. In particolare, i recettori ACE2 sono espressi nel cuore (endotelio delle arterie coronarie, miocliti, fibroblasti, adipociti epicardici), vasi (endoteliali vascolari e cellule lisce), intestino (cellule epiteliali intestinali), polmone (cellule epiteliali tracheali e bronchiali), tipo 2 pneumociti, macrofagi), rene (superficie luminale delle cellule epiteliali tubulari), testicolo, cervello. [13], [14], [15], [16]
Nel polmone, l’ampia superficie delle cellule epiteliali alveolari potrebbe spiegare la vulnerabilità di questo organo alle conseguenze dell’invasione del virus. L’ACE2 è principalmente legato alle membrane cellulari e appena presente nella circolazione solo in forma solubile.
La disintegrina e la metalloproteinasi 17 (ADAM17), sovraregolati dall’angiotensina II attraverso i suoi recettori di tipo 1 (recettori AT1), scindono l’ACE2 ancorato alla membrana, liberando in tal modo una forma attiva circolante di ACE2 con perdita dell’attività catalitica della parte rimanente enzima ancorato alla membrana. [17] Livelli circolanti elevati di ACE2 solubile sono marcatori di diversi stati patologici caratterizzati da una maggiore attività del sistema renina-angiotensina e associati a una prognosi peggiore. [18,19]
ACE2: angeli o diavoli?
Nell’attuale pandemia di SARS-CoV-2, i recettori ACE2 possono essere considerati “diavoli”, essendo la “porta d’ingresso” del virus. Le prove su questo fenomeno sono ora forti e convincenti. Hoffmann et al [2] e Walls et al [3] hanno fornito prove inequivocabili del fatto che SARS-CoV-2 accede alle cellule attraverso i recettori ACE2, come è successo con SARS-Cov. [7]
Altre prove completano il quadro. Ad esempio, in un modello murino di infezione da SARS-CoV, l’ingresso di virus è migliorato dalla sovraespressione di ACE2. [20] Anticorpi anti-ACE2, ma non anticorpi anti-ACE, sono in grado di bloccare l’invasione virale di SARS-Cov, [7] che è anche bloccata da N- (2-aminoetil) -1 aziridina-etamina, un ACE-2 specifico inibitore. [21] Le lesioni polmonari indotte dall’infezione sperimentale SARS-Cov sono meno aggressive nei topi knockout ACE2 rispetto ai topi wild-type. [22]
Allo stesso tempo, tuttavia, i recettori ACE2 esercitano funzioni biologiche salutari che li trasformano in “angeli” sotto diversi aspetti. Una funzione protettiva fondamentale dell’ACE2 è la degradazione dell’angiotensina II in angiotensina 1-7, sebbene l’ACE2 sia in grado di metabolizzare altri peptidi biologici tra cui (des-Arg9) -bradichinina. [15] La degradazione dell’angiotensina II in angiotensina 1-7 è bloccata da inibitori selettivi dell’ACE2 come MLN-4760. [23]
Per comprendere la rilevanza della degradazione dell’angiotensina II da parte dei risultati dell’ACE2, è importante rivedere gli effetti biologici dell’angiotensina II. L’angiotensina II serve non solo come potente vasocostrittore e stimolante del rilascio di aldosterone.
In diversi modelli sperimentali e clinici, l’angiotensina II ha innescato una serie di importanti reazioni avverse, tra cui ipertrofia e disfunzione miocardica, fibrosi interstiziale, disfunzione endoteliale, aumento dell’infiammazione, ipertensione associata all’obesità, stress ossidativo e aumento della coagulazione. [13], [14], [15], [16]
Una discussione dettagliata dei meccanismi attraverso i quali l’angiotensina II media le reazioni di cui sopra è fuori dagli scopi di questa recensione. Nell’attuale pandemia di infezione da SARS-CoV-2 con infiammazione polmonare associata e sindrome da distress respiratorio acuto (ARDS), è interessante notare che l’angiotensina II interferisce anche con l’immunità adattativa attivando i macrofagi [24] e altre cellule del sistema immunitario , con conseguente aumento della produzione di IL-6, [25] TNFα e altri citokynes infiammatori. [26,27]
È importante notare che gli effetti deleteri dell’angiotensina II sintetizzati sopra derivano quasi interamente dalla stimolazione dei recettori AT1. Questa catena di eventi può essere definita come l’asse del recettore ACE → Angiotensin II → AT1.
I recettori ACE2 riducono gli effetti avversi dell’angiotensina II non solo degradando l’angiotensina II, eliminando così o limitando il suo potenziale deleterio, ma anche generando angiotensina1-7.
L’angiotensina 1-7 esercita numerosi effetti salutari e opposti (“controregolatori”) a quelli dell’angiotensina II attraverso un legame efficace con il recettore Mas accoppiato con proteine G e i recettori dell’angiotensina II tipo 2 (recettori AT2). Pertanto, l’asse del recettore ACE2 → Angiotensin1-7 → Mas è contro-regolatorio rispetto all’asse del recettore ACE → Angiotensin II → AT1.
Santos et al. Hanno fornito un’eccellente revisione dei molteplici effetti dell’asse recettore ACE2 → Angiotensin1-7 → Mas. [28]
ACE2 → Angiotensin1-7 → Asse del recettore Mas e polmone
Gli studi sugli effetti polmonari dell’angiotensina 1-7 sembrano particolarmente interessanti. I recettori mas sono espressi sulla superficie delle cellule muscolari lisce bronchiali e dell’epitelio alveolare. [29,30]
Nei modelli sperimentali e clinici di infiammazione polmonare, l’angiotensina 1-7 ha esercitato effetti anti-infiammatori con meno infiltrati di linfociti e neutrofili, riduzione dell’infiammazione perivascolare e peri-bronchiale e prevenzione della successiva fibrosi. [29, [31], [32], [33]
L’ACE2 è espresso sul lato luminale dell’epitelio ciliato bronchiale, dove rimuove un singolo residuo amminoacidico anche dal polipeptide des-Arg [9] bradichinina (DABK), [6] prevenendo così il legame di DABK sul recettore della bradichinina B1 recettore. [34]
In presenza di una ridotta funzione ACE2 nel polmone indotta da endotossine, si verifica un aumento del DABK libero, che a sua volta attiva i recettori B1 con rilascio di citochine pro-infiammatorie e intense infiammazioni e lesioni polmonari. [34]
ACE2 → Angiotensin1-7 → Asse del recettore Mas e trombosi
L’asse ACE2 → Angiotensin1-7 → Mas recettore esercita effetti anti-trombotici [35], [36], [37], [38]. I recettori mas sono espressi su piastrine. [39] La stimolazione dei recettori Mas da parte dell’angiotensina 1-7 aumenta la prostaciclina e il rilascio di NO. [35,36] Il knockout degli animali per i recettori Mas ha un tempo di sanguinamento più breve e una maggiore dimensione dei trombi. [36]
In questi animali, la somministrazione di angiotensina 1-7 induce un marcato effetto antitrombotico che è direttamente correlato ai livelli plasmatici di angiotensina 1-7 [39] ed è inibito da A-779, un antagonista dei recettori di Mas. [35] Pertanto, l’angiotensina 1-7 svolge un ruolo importante nell’opporsi agli effetti pro-trombotici e pro-infiammatori dell’angiotensina II. [40,41]
ACE2 → Angiotensin1-7 → Asse del recettore Mas e sistema endocrino
L’asse del recettore ACE2 → Angiotensin1-7 → Mas è ben espresso nel pancreas dove migliora la secrezione di insulina probabilmente migliorando il flusso sanguigno peri-insulare e inibendo la fibrosi a causa di un maggiore rilascio di NO. [28,42]
I recettori dell’ACE2 sono anche espressi nel tessuto adiposo [43,44] e una riduzione dell’ACE2 è stata osservata nel tessuto adiposo degli animali obesi [44] In esperimenti su animali, le diete ricche di grassi hanno diminuito l’attività dell’ACE2 e l’angiotensina1-7 e aumentato angiotensina II e livelli di pressione sanguigna negli animali maschi, ma non nelle femmine, e queste reazioni sono state inibite dal blocco AT1 con losartan. [45]
Dopo l’ovariectomia, le femmine hanno mostrato reazioni simili a quelle dei maschi. [45] Questi dati suggeriscono che la carenza di ACE2 può favorire l’ipertensione indotta dall’obesità. [45] L’ACE2 è anche espresso negli adipociti cardiaci. [46]
I pazienti obesi con insufficienza cardiaca hanno una maggiore quantità di tessuto adiposo epicardico [46] ed è stato suggerito che la carenza di ACE2 può indurre insufficienza cardiaca con una frazione di eiezione conservata negli animali. [47]
Questo fenomeno è stato attribuito all’infiammazione del tessuto adiposo attraverso l’attivazione locale dei macrofagi, che possiedono recettori AT1 sulla loro membrana cellulare. [26]
Cosa succede all’ACE2 dopo l’associazione SARS-Cov?
SARS-Cov e SARS-CoV2 si legano ai recettori ACE2, con la successiva fusione della membrana e l’ingresso del virus nella cellula, portano a una down-regolazione di questi recettori. [16,22,48] In altri termini, il virus sembra entrare nella cellula insieme al recettore della membrana, che viene rimosso funzionalmente dal sito esterno della membrana.
Di conseguenza, l’asse del recettore ACE → Angiotensina II → Mas viene notevolmente attenuato, con l’amplificazione dell’asse del recettore ACE → Angiotensina II → AT1.
Implicazioni polmonari della down-regolazione ACE2
Poiché l’infiammazione polmonare e la conseguente sindrome da distress respiratorio acuto (ARDS) sono potenzialmente complicazioni mortali di SARS-CoV e SARS-CoV-2, gli studi che affrontano le complicanze polmonari della down-regolazione ACE2 sono della massima importanza. Gli studi che hanno utilizzato diversi modelli di danno polmonare hanno mostrato che la down-regolazione dei recettori ACE2 innesca importanti lesioni infiammatorie nell’albero respiratorio (ispessimento della parete alveolare, edema, infiltrati di cellule infiammatorie, sanguinamento) che sembrano essere mediati dall’angiotensina II. [22, [48], [49], [50]
Instillazione tracheale di fumo di sigaretta, [49] o particolato di diametro aerodinamico inferiore a 2,5 ɥm, [50] induce lesioni polmonari acute con rilascio di citochine infiammatorie IL-6, TNF-α e TGF-β1 e maggiore espressione di ACE, coerente con l’iperattività dell’asse del recettore ACE → Angiotensin II → AT1. [50]
Queste reazioni sono aumentate nei topi knockout ACE2. [50] In un modello di aspirazione acida, che induce una lesione polmonare acuta, le lesioni infiammatorie polmonari erano più gravi e letali negli animali knock-out ACE2. [48]
In questi animali, l’iniezione di ACE2 ricombinante e di bloccanti del recettore AT1 attenua il grado di danno polmonare. [48] Questi risultati suggeriscono fortemente che l’ACE2 protegge dalle lesioni polmonari indotte dall’aspirazione acida. [48]
In particolare, la lesione polmonare è stata indotta dalla proteina virale a punta isolata della SARS-Cov, il ligando per il legame ACE2, in assenza di altri componenti virali. [22] Questo modello ha il merito di studiare l’impatto della down-rule ACE2 in assenza di effetti confondenti di invasione e replicazione virali.
Gli autori hanno scoperto che anche la proteina virale a picco isolata induceva una down-regolazione dei recettori ACE2 con un concomitante aumento dell’angiotensina II nel tessuto polmonare e la precipitazione di gravi lesioni infiammatorie polmonari. [22] Anche in questo modello, i bloccanti dei recettori AT1 hanno attenuato le lesioni polmonari indotte dalla proteina virale di picco. [22]
Un punto chiave da notare è che gli ACE2 sono espressi principalmente in pneumociti di tipo II, piccole cellule cilindriche che rappresentano il 5% di tutti i pneumociti. [51] I pneumociti di tipo 2 sono responsabili della produzione di tensioattivo alveolare e allo stesso tempo funzionano come cellule staminali, progenitori dei pneumociti di tipo I (95% di tutti i pneumociti) che sono responsabili degli scambi di gas. [52]
Pertanto, il danno dei pneumociti di tipo II dovuto al legame del coronavirus ai recettori ACE2 è devastante per almeno tre motivi: 1) iperattività dell’asse del recettore ACE → Angiotensin II → AT1 locale non opposto; 2) ridotta produzione di tensioattivo alveolare da parte di pneumociti feriti di tipo II con conseguente riduzione dell’elasticità polmonare; 3) riduzione della riparazione dei pneumociti di tipo I con conseguente alterazione degli scambi di gas e fibrosi. [53]
Caratteristiche cliniche dei pazienti infetti da SARS-CoV e SARS-CoV-2
Studi provenienti dalla Cina e dall’Italia hanno dimostrato che l’ipertensione, il diabete e la storia di malattie cardiovascolari sono le comorbilità più frequenti nei pazienti con infezione da SARS-CoV-2. [54], [55], [56] L’età avanzata e il sesso maschile sono altri due fattori associati all’infezione da SARS-CoV-2. [54], [55], [56] Un’immagine simile è emersa alcuni anni fa con l’infezione SARS-CoV. [57,58]
In uno studio condotto su 201 pazienti con infezione da SARS-CoV-2, la maggior parte dei pazienti erano uomini (63,7% dei pazienti), l’età media era di 51 anni e le comorbidità più frequenti erano ipertensione (19,4%), diabete (10,9%) e storia di malattie cardiovascolari (4.0). [55] In particolare, i pazienti che hanno sviluppato ARDS erano più anziani e avevano una maggiore prevalenza di ipertensione (27,4% contro 13,7%), diabete (19,0% contro 5,1%) rispetto a quelli che non avevano sviluppato ARDS. [55]
In un’analisi multivariata, i fattori associati alla progressione dalla sindrome ARDS alla morte includevano l’età avanzata, la neutrofilia e l’iper-coagulazione, principalmente riflesse da un D-dimero più elevato. [55]
Parametri di coagulazione anormali e trombosi migliorata predicono una prognosi sfavorevole nei pazienti con SARS-CoV-2. [59] Una meta-analisi di 8 studi condotti in Cina su un totale di 46.248 pazienti infetti da SARS-CoV2 ha confermato che l’ipertensione, il diabete e la storia di malattie cardiovascolari erano le comorbilità più frequenti in questi pazienti. [56] Ancora una volta, l’ipertensione e la storia di malattie cardiovascolari erano significativamente più prevalenti tra i pazienti più gravi. [56]
In una recente analisi di 1591 pazienti infetti dall’Italia, l’età media dei pazienti era di 63 anni, gli uomini erano dell’82% e la prevalenza di pazienti con ipertensione, diabete e precedenti malattie cardiovascolari era rispettivamente del 49%, 17% e 21%. [54]
I pazienti con ipertensione erano più anziani di quelli senza ipertensione (66 vs 62 anni, p = 0,005). Confrontando i pazienti deceduti nel reparto di terapia intensiva con quelli sopravvissuti, i primi erano più anziani e presentavano una prevalenza maggiore di ipertensione (63% vs. 40%, p <0,001). [54]
Carenza di ACE2: un ruolo centrale nell’infezione SARS-CoV-2?
È interessante notare che diverse condizioni associate all’infezione virale e alla gravità della malattia condividono un grado variabile di carenza di ACE2. Ad esempio, l’espressione dell’ACE2 nei polmoni diminuisce notevolmente con l’invecchiamento, [60] in misura maggiore negli uomini rispetto alle donne. [60] Il diabete mellito è stato associato a una ridotta espressione di ACE2, probabilmente come effetto della glicosilazione. [61], [62], [63]
Diversi studi sperimentali e clinici indicano che la carenza di ACE2 ottenuta attraverso la cancellazione o l’inibizione può essere un fattore causale per l’ipertensione. [14,64] Il trattamento con ACE2 ricombinante solubile riduce l’aumento della pressione sanguigna provocato dall’angiotensina II, aumenta l’angiotensina1-7 e riduce l’angiotensina II. [65]
La carenza di ACE2 è stata associata all’esacerbazione dell’ipertensione e dell’ipertrofia cardiaca indotta dall’angiotensina II, [66] e dal rimodellamento ventricolare sinistro disadattivo dopo infarto miocardico. [67]
Inoltre, la carenza di ACE2 aumenta la suscettibilità all’insufficienza cardiaca. [14] Una perdita di eterozigoti di ACE2 è ritenuta sufficiente per aumentare la suscettibilità alle malattie cardiache. [68]
Date le premesse di cui sopra, si è tentati di ipotizzare (Figura 2) che la carenza di ACE2 possa svolgere un ruolo centrale nella patogenesi dell’infezione da SARS-CoV-2. La sottoregolazione dell’ACE2 indotta dall’invasione virale potrebbe essere particolarmente dannosa nei soggetti con deficit ACE2 al basale dovuto, ad esempio, all’età avanzata, al diabete, all’ipertensione e alle precedenti malattie cardiache inclusa l’insufficienza cardiaca.
Ulteriori informazioni: Iziah E Sama et al, Concentrazioni plasmatiche circolanti dell’enzima 2 di conversione dell’angiotensina in uomini e donne con insufficienza cardiaca ed effetti degli inibitori della renina-angiotensina-aldosterone, European Heart Journal (2020). DOI: 10.1093 / eurheartj / ehaa373
References
- Chan-Yeung M, Xu RH. SARS: epidemiology. Respirology. 2003;8(Suppl:S9-14)
- Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, Muller MA, Drosten C, Pohlmann S. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020 doi: 10.1016/j.cell.2020.02.052.
- Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020 doi: 10.1016/j.cell.2020.02.058.
- Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275:33238–33243.
- Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, Breitbart RE, Acton S. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res. 2000;87:E1–E9.
- Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, Godbout K, Parsons T, Baronas E, Hsieh F, Acton S, Patane M, Nichols A, Tummino P. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem. 2002;277:14838–14843
- Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454.
- Xu X, Chen P, Wang J, Feng J, Zhou H, Li X, Zhong W, Hao P. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci China Life Sci. 2020;63:457–460.
- Hussain M, Jabeen N, Raza F, Shabbir S, Baig AA, Amanullah A, Aziz B. Structural Variations in Human ACE2 may Influence its Binding with SARS-CoV-2 Spike Protein. J Med Virol. 2020 doi: 10.1002/jmv.25832.
- Glowacka I, Bertram S, Muller MA, Allen P, Soilleux E, Pfefferle S, Steffen I, Tsegaye TS, He Y, Gnirss K, Niemeyer D, Schneider H, Drosten C, Pohlmann S. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J Virol. 2011;85:4122–4134.
- Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, Oliveira-dos-Santos AJ, da Costa J, Zhang L, Pei Y, Scholey J, Ferrario CM, Manoukian AS, Chappell MC, Backx PH, Yagil Y, Penninger JM. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822–828.
- Batlle D, Wysocki J, Satchell K. Soluble angiotensin-converting enzyme 2: a potential approach for coronavirus infection therapy? Clin Sci (Lond) 2020;134:543–545.
- Kuba K, Imai Y, Penninger JM. Multiple functions of angiotensin-converting enzyme 2 and its relevance in cardiovascular diseases. Circ J. 2013;77:301–308.
- Patel VB, Zhong JC, Grant MB, Oudit GY. Role of the ACE2/Angiotensin 1-7 Axis of the Renin-Angiotensin System in Heart Failure. Circ Res. 2016;118:1313–1326.
- Turner AJ, Hiscox JA, Hooper NM. ACE2: from vasopeptidase to SARS virus receptor. Trends Pharmacol Sci. 2004;25:291–294.
- Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020 doi: 10.1007/s00134-020-05985-9.
- Xu J, Sriramula S, Xia H, Moreno-Walton L, Culicchia F, Domenig O, Poglitsch M, Lazartigues E. Clinical Relevance and Role of Neuronal AT1 Receptors in ADAM17-Mediated ACE2 Shedding in Neurogenic Hypertension. Circ Res. 2017;121:43–55.
- Bitker L, Burrell LM. Classic and Nonclassic Renin-Angiotensin Systems in the Critically Ill. Crit Care Clin. 2019;35:213–227.
- Wysocki J, Goodling A, Burgaya M, Whitlock K, Ruzinski J, Batlle D, Afkarian M. Urine RAS components in mice and people with type 1 diabetes and chronic kidney disease. Am J Physiol Renal Physiol. 2017;313:F487–F494.
- Yang XH, Deng W, Tong Z, Liu YX, Zhang LF, Zhu H, Gao H, Huang L, Liu YL, Ma CM, Xu YF, Ding MX, Deng HK, Qin C. Mice transgenic for human angiotensin-converting enzyme 2 provide a model for SARS coronavirus infection. Comp Med. 2007;57:450–459.
- Huentelman MJ, Zubcevic J, Hernandez Prada JA, Xiao X, Dimitrov DS, Raizada MK, Ostrov DA. Structure-based discovery of a novel angiotensin-converting enzyme 2 inhibitor. Hypertension. 2004;44:903–906.
- Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, Huan Y, Yang P, Zhang Y, Deng W, Bao L, Zhang B, Liu G, Wang Z, Chappell M, Liu Y, Zheng D, Leibbrandt A, Wada T, Slutsky AS, Liu D, Qin C, Jiang C, Penninger JM. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11:875–879.
- Trask AJ, Averill DB, Ganten D, Chappell MC, Ferrario CM. Primary role of angiotensin-converting enzyme-2 in cardiac production of angiotensin-(1-7) in transgenic Ren-2 hypertensive rats. Am J Physiol Heart Circ Physiol. 2007;292:H3019–H3024.
- Bernstein KE, Khan Z, Giani JF, Cao DY, Bernstein EA, Shen XZ. Angiotensin-converting enzyme in innate and adaptive immunity. Nat Rev Nephrol. 2018;14:325–336.
- Recinos A, 3rd, LeJeune WS, Sun H, Lee CY, Tieu BC, Lu M, Hou T, Boldogh I, Tilton RG, Brasier AR. Angiotensin II induces IL-6 expression and the Jak-STAT3 pathway in aortic adventitia of LDL receptor-deficient mice. Atherosclerosis. 2007;194:125–133.
- Yamamoto S, Yancey PG, Zuo Y, Ma LJ, Kaseda R, Fogo AB, Ichikawa I, Linton MF, Fazio S, Kon V. Macrophage polarization by angiotensin II-type 1 receptor aggravates renal injury-acceleration of atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:2856–2864.
- Lee YB, Nagai A, Kim SU. Cytokines, chemokines, and cytokine receptors in human microglia. J Neurosci Res. 2002;69:94–103.
- Santos RAS, Sampaio WO, Alzamora AC, Motta-Santos D, Alenina N, Bader M, Campagnole-Santos MJ. The ACE2/Angiotensin-(1-7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1-7) Physiol Rev. 2018;98:505–553.
- Magalhaes GS, Rodrigues-Machado MG, Motta-Santos D, Silva AR, Caliari MV, Prata LO, Abreu SC, Rocco PR, Barcelos LS, Santos RA, Campagnole-Santos MJ. Angiotensin-(1-7) attenuates airway remodelling and hyperresponsiveness in a model of chronic allergic lung inflammation. Br J Pharmacol. 2015;172:2330–2342.
- El-Hashim AZ, Renno WM, Raghupathy R, Abduo HT, Akhtar S, Benter IF. Angiotensin-(1-7) inhibits allergic inflammation, via the MAS1 receptor, through suppression of ERK1/2- and NF-kappaB-dependent pathways. Br J Pharmacol. 2012;166:1964–1976.
- Chen Q, Yang Y, Huang Y, Pan C, Liu L, Qiu H. Angiotensin-(1-7) attenuates lung fibrosis by way of Mas receptor in acute lung injury. J Surg Res. 2013;185:740–747.
- Li Y, Cao Y, Zeng Z, Liang M, Xue Y, Xi C, Zhou M, Jiang W. Angiotensin-converting enzyme 2/angiotensin-(1-7)/Mas axis prevents lipopolysaccharide-induced apoptosis of pulmonary microvascular endothelial cells by inhibiting JNK/NF-kappaB pathways. Sci Rep. 2015;5:8209.
- Meng Y, Yu CH, Li W, Li T, Luo W, Huang S, Wu PS, Cai SX, Li X. Angiotensin-converting enzyme 2/angiotensin-(1-7)/Mas axis protects against lung fibrosis by inhibiting the MAPK/NF-kappaB pathway. Am J Respir Cell Mol Biol. 2014;50:723–736.
- Sodhi CP, Wohlford-Lenane C, Yamaguchi Y, Prindle T, Fulton WB, Wang S, McCray PB, Jr., Chappell M, Hackam DJ, Jia H. Attenuation of pulmonary ACE2 activity impairs inactivation of des-Arg(9) bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am J Physiol Lung Cell Mol Physiol. 2018;314:L17–L31.
- Fang C, Stavrou E, Schmaier AA, Grobe N, Morris M, Chen A, Nieman MT, Adams GN, LaRusch G, Zhou Y, Bilodeau ML, Mahdi F, Warnock M, Schmaier AH. Angiotensin 1-7 and Mas decrease thrombosis in Bdkrb2-/- mice by increasing NO and prostacyclin to reduce platelet spreading and glycoprotein VI activation. Blood. 2013;121:3023–3032.
- Fraga-Silva RA, Pinheiro SV, Goncalves AC, Alenina N, Bader M, Santos RA. The antithrombotic effect of angiotensin-(1-7) involves mas-mediated NO release from platelets. Mol Med. 2008;14:28–35.
- Kucharewicz I, Pawlak R, Matys T, Pawlak D, Buczko W. Antithrombotic effect of captopril and losartan is mediated by angiotensin-(1-7) Hypertension. 2002;40:774–779.
- Pai WY, Lo WY, Hsu T, Peng CT, Wang HJ. Angiotensin-(1-7) Inhibits Thrombin-Induced Endothelial Phenotypic Changes and Reactive Oxygen Species Production via NADPH Oxidase 5 Downregulation. Front Physiol. 2017;8:994.
- Fraga-Silva RA, Costa-Fraga FP, De Sousa FB, Alenina N, Bader M, Sinisterra RD, Santos RA. An orally active formulation of angiotensin-(1-7) produces an antithrombotic effect. Clinics (Sao Paulo) 2011;66:837–841.
- Liang B, Wang X, Zhang N, Yang H, Bai R, Liu M, Bian Y, Xiao C, Yang Z. Angiotensin-(1-7) Attenuates Angiotensin II-Induced ICAM-1, VCAM-1, and MCP-1 Expression via the MAS Receptor Through Suppression of P38 and NF-kappaB Pathways in HUVECs. Cell Physiol Biochem. 2015;35:2472–2482.
- Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82–C97.
- Yuan L, Li Y, Li G, Song Y, Gong X. Ang(1-7) treatment attenuates beta-cell dysfunction by improving pancreatic microcirculation in a rat model of Type 2 diabetes. J Endocrinol Invest. 2013;36:931–937.
- Gembardt F, Sterner-Kock A, Imboden H, Spalteholz M, Reibitz F, Schultheiss HP, Siems WE, Walther T. Organ-specific distribution of ACE2 mRNA and correlating peptidase activity in rodents. Peptides. 2005;26:1270–1277.
- Gupte M, Boustany-Kari CM, Bharadwaj K, Police S, Thatcher S, Gong MC, English VL, Cassis LA. ACE2 is expressed in mouse adipocytes and regulated by a high-fat diet. Am J Physiol Regul Integr Comp Physiol. 2008;295:R781–R788.
- Gupte M, Thatcher SE, Boustany-Kari CM, Shoemaker R, Yiannikouris F, Zhang X, Karounos M, Cassis LA. Angiotensin converting enzyme 2 contributes to sex differences in the development of obesity hypertension in C57BL/6 mice. Arterioscler Thromb Vasc Biol. 2012;32:1392–1399.
- Patel VB, Basu R, Oudit GY. ACE2/Ang 1-7 axis: A critical regulator of epicardial adipose tissue inflammation and cardiac dysfunction in obesity. Adipocyte. 2016;5:306–311.
- Patel VB, Mori J, McLean BA, Basu R, Das SK, Ramprasath T, Parajuli N, Penninger JM, Grant MB, Lopaschuk GD, Oudit GY. ACE2 Deficiency Worsens Epicardial Adipose Tissue Inflammation and Cardiac Dysfunction in Response to Diet-Induced Obesity. Diabetes. 2016;65:85–95.
- Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, Yang P, Sarao R, Wada T, Leong-Poi H, Crackower MA, Fukamizu A, Hui CC, Hein L, Uhlig S, Slutsky AS, Jiang C, Penninger JM. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436:112–116.
- Hung YH, Hsieh WY, Hsieh JS, Liu FC, Tsai CH, Lu LC, Huang CY, Wu CL, Lin CS. Alternative Roles of STAT3 and MAPK Signaling Pathways in the MMPs Activation and Progression of Lung Injury Induced by Cigarette Smoke Exposure in ACE2 Knockout Mice. Int J Biol Sci. 2016;12:454–465.
- Lin CI, Tsai CH, Sun YL, Hsieh WY, Lin YC, Chen CY, Lin CS. Instillation of particulate matter 2.5 induced acute lung injury and attenuated the injury recovery in ACE2 knockout mice. Int J Biol Sci. 2018;14:253–265.
- Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631–637.
- Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, Randell SH, Noble PW, Hogan BL. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. 2013;123:3025–3036.
- Rivellese F, Prediletto E. ACE2 at the centre of COVID-19 from paucisymptomatic infections to severe pneumonia. Autoimmun Rev. 2020 doi: 10.1016/j.autrev.2020.102536:102536.
- Grasselli G, Zangrillo A, Zanella A, Antonelli M, Cabrini L, Castelli A, Cereda D, Coluccello A, Foti G, Fumagalli R, Iotti G, Latronico N, Lorini L, Merler S, Natalini G, Piatti A, Ranieri MV, Scandroglio AM, Storti E, Cecconi M, Pesenti A, Network C-LI. Baseline Characteristics and Outcomes of 1591 Patients Infected With SARS-CoV-2 Admitted to ICUs of the Lombardy Region, Italy. JAMA. 2020 doi: 10.1001/jama.2020.5394.
- Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, Huang H, Zhang L, Zhou X, Du C, Zhang Y, Song J, Wang S, Chao Y, Yang Z, Xu J, Zhou X, Chen D, Xiong W, Xu L, Zhou F, Jiang J, Bai C, Zheng J, Song Y. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern Med. 2020 doi: 10.1001/jamainternmed.2020.0994.
- Yang J, Zheng Y, Gou X, Pu K, Chen Z, Guo Q, Ji R, Wang H, Wang Y, Zhou Y. Prevalence of comorbidities in the novel Wuhan coronavirus (COVID-19) infection: a systematic review and meta-analysis. Int J Infect Dis. 2020 doi: 10.1016/j.ijid.2020.03.017.
- Booth CM, Matukas LM, Tomlinson GA, Rachlis AR, Rose DB, Dwosh HA, Walmsley SL, Mazzulli T, Avendano M, Derkach P, Ephtimios IE, Kitai I, Mederski BD, Shadowitz SB, Gold WL, Hawryluck LA, Rea E, Chenkin JS, Cescon DW, Poutanen SM, Detsky AS. Clinical features and short-term outcomes of 144 patients with SARS in the greater Toronto area. JAMA. 2003;289:2801–2809.
- Chan JW, Ng CK, Chan YH, Mok TY, Lee S, Chu SY, Law WL, Lee MP, Li PC. Short term outcome and risk factors for adverse clinical outcomes in adults with severe acute respiratory syndrome (SARS) Thorax. 2003;58:686–689.
- Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020;18:844–847.
- Xie X, Chen J, Wang X, Zhang F, Liu Y. Age- and gender-related difference of ACE2 expression in rat lung. Life Sci. 2006;78:2166–2171.
- Pal R, Bhansali A. COVID-19, Diabetes Mellitus and ACE2: The conundrum. Diabetes Res Clin Pract. 2020 doi: 10.1016/j.diabres.2020.108132:108132.
- Tikellis C, Thomas MC. Angiotensin-Converting Enzyme 2 (ACE2) Is a Key Modulator of the Renin Angiotensin System in Health and Disease. Int J Pept. 2012;2012
- Yamagata R, Nemoto W, Nakagawasai O, Takahashi K, Tan-No K. Downregulation of spinal angiotensin converting enzyme 2 is involved in neuropathic pain associated with type 2 diabetes mellitus in mice. Biochem Pharmacol. 2020;174
- Patel SK, Velkoska E, Freeman M, Wai B, Lancefield TF, Burrell LM. From gene to protein-experimental and clinical studies of ACE2 in blood pressure control and arterial hypertension. Front Physiol. 2014;5:227.
- Wysocki J, Ye M, Rodriguez E, Gonzalez-Pacheco FR, Barrios C, Evora K, Schuster M, Loibner H, Brosnihan KB, Ferrario CM, Penninger JM, Batlle D. Targeting the degradation of angiotensin II with recombinant angiotensin-converting enzyme 2: prevention of angiotensin II-dependent hypertension. Hypertension. 2010;55:90–98.
- Zhong J, Basu R, Guo D, Chow FL, Byrns S, Schuster M, Loibner H, Wang XH, Penninger JM, Kassiri Z, Oudit GY. Angiotensin-converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis, and cardiac dysfunction. Circulation. 2010;122:717–728. 718 p following 728.
- Kassiri Z, Zhong J, Guo D, Basu R, Wang X, Liu PP, Scholey JW, Penninger JM, Oudit GY. Loss of angiotensin-converting enzyme 2 accelerates maladaptive left ventricular remodeling in response to myocardial infarction. Circ Heart Fail. 2009;2:446–455
- Wang W, Patel VB, Parajuli N, Fan D, Basu R, Wang Z, Ramprasath T, Kassiri Z, Penninger JM, Oudit GY. Heterozygote loss of ACE2 is sufficient to increase the susceptibility to heart disease. J Mol Med (Berl) 2014;92:847–858.
- Mehta P, MaAuley DF, Brown M, Sanchez E, Tattersall RS, Manson J. COVID-19: consider cytokine storm syndromes and immunosuppression. The Lancet. 2020;395:1–2
- Akhmerov A, Marban E. COVID-19 and the Heart. Circ Res. 2020 doi: 10.1161/CIRCRESAHA.120.317055.
- Peiro C, Moncada S. Substituting Angiotensin-(1-7) to Prevent Lung Damage in SARSCoV2 Infection? Circulation. 2020 doi: 10.1161/CIRCULATIONAHA.120.047297.
- Ferrario CM, Jessup J, Chappell MC, Averill DB, Brosnihan KB, Tallant EA, Diz DI, Gallagher PE. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. 2005;111:2605–2610.
- Gallagher PE, Ferrario CM, Tallant EA. MAP kinase/phosphatase pathway mediates the regulation of ACE2 by angiotensin peptides. Am J Physiol Cell Physiol. 2008;295:C1169–C1174.
- Ishiyama Y, Gallagher PE, Averill DB, Tallant EA, Brosnihan KB, Ferrario CM. Upregulation of angiotensin-converting enzyme 2 after myocardial infarction by blockade of angiotensin II receptors. Hypertension. 2004;43:970–976. [PubMed]
- Jessup JA, Gallagher PE, Averill DB, Brosnihan KB, Tallant EA, Chappell MC, Ferrario CM. Effect of angiotensin II blockade on a new congenic model of hypertension derived from transgenic Ren-2 rats. Am J Physiol Heart Circ Physiol. 2006;291:H2166–H2172.
- Bavishi C, Maddox TM, Messerli FH. Coronavirus Disease 2019 (COVID-19) Infection and Renin Angiotensin System Blockers. JAMA Cardiol. 2020 doi: 10.1001/jamacardio.2020.1282.
- Danser AHJ, Epstein M, Batlle D. Renin-Angiotensin System Blockers and the COVID-19 Pandemic: At Present There Is No Evidence to Abandon Renin-Angiotensin System Blockers. Hypertension. 2020 doi:10.1161/HYPERTENSIONAHA.120.15082:HYPERTENSIONAHA12015082.
- Esler M, Esler D. Can angiotensin receptor-blocking drugs perhaps be harmful in the COVID-19 pandemic? J. Hypertens. 2020;38:1–2.
- Fang L, Karakiulakis G, TRoth M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir Med. 2020 doi: 10.1016/S2213-2600(20)30116-8:1.
- Gurwitz D. Angiotensin receptor blockers as tentative SARS-CoV-2 therapeutics. Drug Dev Res. 2020 doi: 10.1002/ddr.21656.
- Kuster GM, Pfister O, Burkard T, Zhou Q, Twerenbold R, Haaf P, Widmer AF, Osswald S. SARS-CoV2: should inhibitors of the renin-angiotensin system be withdrawn in patients with COVID-19? Eur Heart J. 2020 doi: 10.1093/eurheartj/ehaa235.
- Vaduganathan M, Vardeny O, Michel T, McMurray JJV, Pfeffer MA, Solomon SD. Renin-Angiotensin-Aldosterone System Inhibitors in Patients with Covid-19. N Engl J Med. 2020 doi: 10.1056/NEJMsr2005760.
- Verdecchia P, Angeli F, Reboldi G. Angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers and coronavirus. J Hypertens. 2020;38 (in press)
- Verdecchia P, Reboldi G, Cavallini C, Mazzotta G, Angeli F. ACE-inibitori, sartani e sindrome respiratoria acuta da coronavirus 2. G Ital Cardiol (Rome) 2020;21:1–7.







{kind=link}