











 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Scientists from USC and the IFOM Cancer Institute in Milan have found that a fasting-mimicking diet could be more effective at treating some types of cancer when combined with vitamin C.
In studies on mice, researchers found that the combination delayed tumor progression in multiple mouse models of colorectal cancer; in some mice, it caused disease regression. The results were published in the journal Nature Communications.
“For the first time, we have demonstrated how a completely non-toxic intervention can effectively treat an aggressive cancer,” said Valter Longo, the study senior author and the director of the USC Longevity Institute at the USC Leonard Davis School of Gerontology and professor of biological sciences at the USC Dornsife College of Letters, Arts and Sciences. “We have taken two treatments that are studied extensively as interventions to delay aging— a fasting-mimicking diet and vitamin C – and combined them as a powerful treatment for cancer.”
The researchers said that while fasting remains a challenging option for cancer patients, a safer, more feasible option is a low-calorie, plant-based diet that causes cells to respond as if the body were fasting.
Their findings suggest that a low-toxicity treatment of fasting-mimicking diet plus vitamin C has the potential to replace more toxic treatments.
Results of prior research on the cancer-fighting potential of vitamin C have been mixed. Recent studies, though, are beginning to show some efficacy, especially in combination with chemotherapy.
In this new study, the research team wanted to find out whether a fasting-mimicking diet could enhance the high-dose vitamin C tumor-fighting action by creating an environment that would be unsustainable for cancer cells but still safe for normal cells.
“Our first in vitro experiment showed remarkable effects,” said Longo.
“When used alone, fasting-mimicking diet or vitamin C alone reduced cancer cell growth and caused a minor increase in cancer cell death. But when used together, they had a dramatic effect, killing almost all cancerous cells.”
Longo and his colleagues detected this strong effect only in cancer cells that had a mutation that is regarded as one of the most challenging targets in cancer research.
These mutations in the KRAS gene signal the body is resisting most cancer-fighting treatments, and they reduce a patient’s survival rate. KRAS mutations occur in approximately a quarter of all human cancers and are estimated to occur in up to half of all colorectal cancers.
The study also provided clues about why previous studies of vitamin C as a potential anticancer therapy showed limited efficacy. By itself, a vitamin C treatment appears to trigger the KRAS-mutated cells to protect cancer cells by increasing levels of ferritin, a protein that binds iron.
But by reducing levels of ferritin, the scientists managed to increase vitamin C’s toxicity for the cancer cells. Amid this finding, the scientists also discovered that colorectal cancer patients with high levels of the iron-binding protein have a lower chance of survival.
“In this study, we observed how fasting-mimicking diet cycles are able to increase the effect of pharmacological doses of vitamin C against KRAS-mutated cancers,” said Maira Di Tano, a study co-author at the IFOM, FIRC Institute of Molecular Oncology in Milan, Italy.
“This occurs through the regulation of the levels of iron and of the molecular mechanisms involved in oxidative stress. The results particularly pointed to a gene that regulates iron levels: heme-oxygenase-1.”
The research team’s prior studies showed that fasting and a fasting-mimicking diet slow cancer’s progression and make chemotherapy more effective in tumor cells, while protecting normal cells from chemotherapy-associated side effects.
The combination enhances the immune system’s anti-tumor response in breast cancer and melanoma mouse models.
The scientists believe cancer will eventually be treated with low-toxicity drugs in a manner similar to how antibiotics are used to treat infections that kill particular bacteria, but which can be substituted by other drugs if the first is not effective.
To move toward that goal, they say they needed to first test two hypotheses: that their non-toxic combination interventions would work in mice, and that it would look promising for human clinical trials. In this new study, they said that they’ve demonstrated both.
At least five clinical trials, including one at USC on breast cancer and prostate cancer patients, are now investigating the effects of the fasting-mimicking diets in combination with different cancer-fighting drugs.
FMD enhances vitamin C toxicity in KRAS-mutant cancer cells
We investigated whether the fasting/FMD potentiates the anti-cancer effect of vitamin C against different KRAS-mutant cancer models. Human (HCT116, DLD-1) and murine (CT26) KRAS-mutant CRC cell lines, as well as KRAS-mutant lung cancer (H23, H727) and pancreatic ductal adenocarcinoma (PANC1) cells, were grown in control medium (1 g/L glucose and 10% serum; CTR) or in a FMD-like medium (0.5 g/L glucose and 1% serum), here referred to as short-term starvation condition (STS), which mimics the reduction of extracellular glucose and growth factor concentrations that occurs during prolonged (>48 h) fasting or FMD in vivo, with or without pharmacological concentrations of vitamin C (≥0.3 mM).
Consistent with recent findings4, KRAS-mutant tumor cells were more susceptible to vitamin C compared with KRAS-wild-type cancer cells (Fig. 1a, b). When cancer cells were grown under STS conditions before and during treatment, vitamin C-mediated toxicity was strongly enhanced (Fig. 1a).
Conversely, KRAS-wild-type CRC (SW48, HT29), prostate cancer (PC-3), ovarian cancer (COV362) cell lines and a normal colon cell line (CCD841CoN) were resistant to vitamin C when used both as a single agent and in combination with STS (Fig. 1b).
In agreement with the selective toxicity of STS + vitamin C in KRAS-mutant tumor cells, we found that HT29 and SW48 cells genetically modified to express the active form of KRAS were more susceptible to STS + vitamin C compared with their wild-type isogenic counterpart (Fig. 1c and Supplementary Fig. 1).
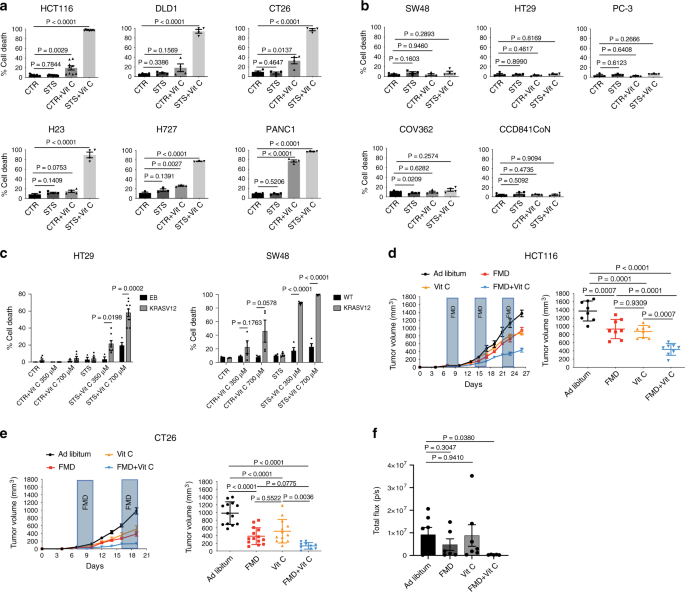
Consistent with our in vitro results, we found that FMD cycles combined with daily vitamin C treatment (4 g per kg twice a day) were effective in delaying the progression of KRAS mutated tumors in different mouse models (Fig. 1d–f). In particular, weekly cycles of a three days FMD were sufficient to reduce KRAS mutated tumor growth to the same extent as high-dose vitamin C (Fig. 1d, e). Notably, weekly FMD and daily vitamin C showed the best therapeutic outcome in reducing CRC progression in xenograft and syngeneic mouse models as well as in an orthotopic model (Fig. 1d–f and Supplementary Fig. 2a).
Furthermore, the FMD-vitamin C combination was safe and well tolerated in both mouse strains, as indicated by mouse body weight loss, which did not exceed 20% and was rapidly recovered upon refeeding (Supplementary Fig. 2b).
ROS mediate sensitization to vitamin C
We previously showed that fasting/FMD sensitizes different types of cancer cells to chemotherapy through a mechanism that involves increased ROS production17,25. ROS, including H2O2 and superoxide, generated as by-products of normal metabolism, cause damage to DNA, lipids and proteins26.
Recent studies have shown that KRAS mutations promote metabolic reprogramming to sustain high-proliferation rates, accompanied by a higher oxidative state compared with KRAS-wild-type cells11,12,27,28. Thus, we hypothesized that the higher oxidative state of KRAS-mutant tumors may underlie the selective mechanism of FMD + vitamin C toxicity.
Notably, the combination of STS and vitamin C strongly induced DNA damage in CT26 and HCT116 cells, as indicated by the phosphorylation of the histone H2AX (Fig. 2a), suggesting that oxidative stress may participate in mediating this cytotoxic effect. Indeed, the combination of STS and vitamin C selectively exacerbated ROS production in KRAS-mutant tumor cells (Fig. 2b and Supplementary Fig. 3a).
Consistent with the vitamin C-dependent production of H2O2, we observed the same selective increase in ROS in KRAS-mutant cancer cells grown in STS condition and treated with H2O2 (Supplementary Fig. 3b).
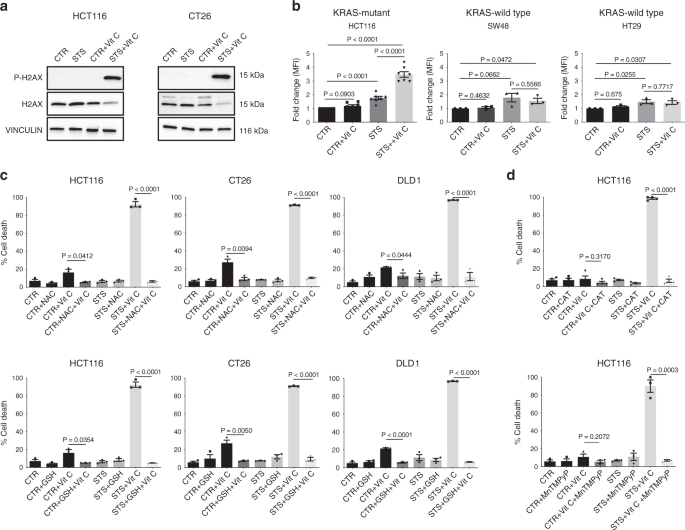
To directly assess whether increased ROS production is a causative or a secondary event in STS-induced sensitization to vitamin C, we evaluated the effect of different antioxidants on STS + vitamin C toxicity (Fig. 2c, d and Supplementary Fig. 3c). Glutathione (GSH) and N-acetyl cysteine (NAC), as well as prior exposure to membrane-impermeable catalase (CAT) or membrane-permeable superoxide dismutase (SOD)/catalase mimetic MnTMPyP were able to revert the STS + vitamin C induced cell death (Fig. 2c, d).
Collectively, these findings indicate that ROS production and redox alterations represents a central mechanism through which the STS and vitamin C combination selectively kills KRAS mutated cancer cells.
Iron is involved in FMD-mediated toxicity
A large body of evidence shows that the mechanism underlying vitamin C’s anti-cancer effects relies on H2O2 production and that the LIP plays a fundamental role in this process3,6,7.
In the presence of free iron, high H2O2 levels have pro-oxidant effects in part through the generation of hydroxyl radicals via Fenton reaction and the induction of oxidative damage3,7.
Since the combination of FMD/STS and vitamin C increased ROS levels in KRAS-mutant cancer cells, we investigated whether this correlates with an increased pool of labile ferrous iron. In HCT116 cells, ferrous ion (Fe2+) levels were significantly increased upon STS and vitamin C co-treatment compared with all other conditions (Fig. 3a), suggesting a potential role of iron in mediating this effect.
Ferritin, the main protein involved in iron binding and storage, regulates the intracellular LIP and its downregulation has been shown to increase LIP in KRAS-mutant cancer cells10,11,12.
Thus, we measured the levels of the heavy subunit of ferritin (FTH), which is responsible for the iron storage through its ferroxidase activity10. Consistent with the increase in ferrous iron, we found that STS, alone or in combination with vitamin C, downregulated FTH protein expression selectively in KRAS-mutant cancer cells. On the other hand, vitamin C reversed STS-induced ferritin downregulation in KRAS-wild-type tumor cells (Fig. 3b, Supplementary Fig. 4).
These in vitro results were also confirmed in vivo, where FMD cycles combined with vitamin C treatment downregulated FTH protein expression in HCT116-derived tumors (Fig. 3c).
Fig. 3: Iron is involved in FMD + vitamin C toxicity selectively in KRAS mutated cancer cells.
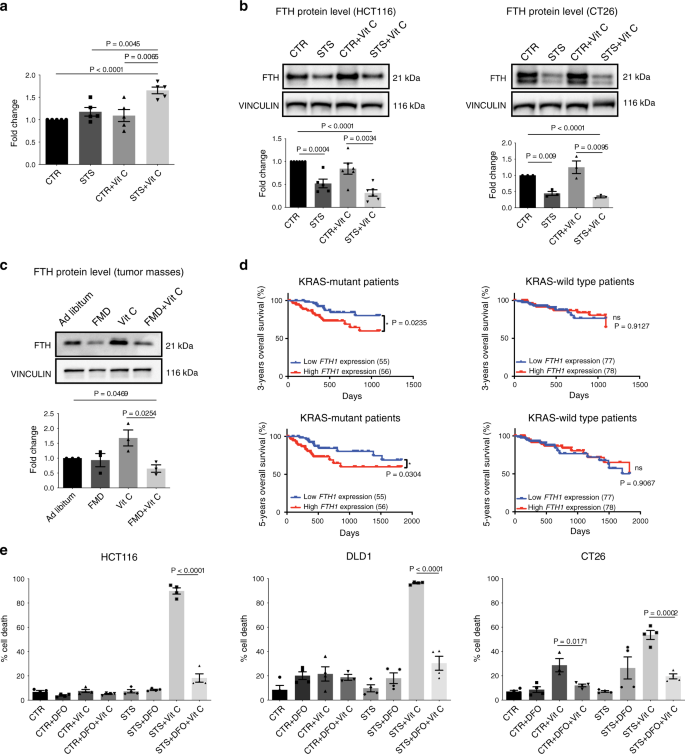
Consistent with a sensitizing effect of FTH downregulation in KRAS mutant cancer cells, our analysis of CRC patient-survival data acquired from The Cancer Genome Atlas Database (TCGA), showed that patients with KRAS mutated tumors and low intratumoral ferritin transcriptional level had a longer 3- and 5-year overall survival when compared with patients whose tumors expressed high ferritin level (Fig. 3d). This association, which was not observed for wild type KRAS tumors, supports the role for FMD + vitamin C as a strategy to maintain low ferritin levels and increase ROS to treat KRAS mutated tumors. Notably, these data represent less than 200 patients; therefore, analyses of larger patient populations are required to better understand the role of ferritin expression, or activity, in KRAS mutated tumors progression.
To assess whether the alteration in cellular iron content contributes to STS-mediated sensitization to vitamin C, KRAS-mutant CRC cells grown in STS conditions were treated with the iron chelator desferrioxamine (DFO) before vitamin C exposure. Consistent with our hypothesis, DFO treatment preceding vitamin C exposure rescued vitamin C-induced cell cytotoxicity (Fig. 3e), thus confirming that the increase in intracellular free iron mediated by FMD/STS and vitamin C is, at least in part, responsible for their cytotoxic effect.
FMD reverses the effect of vitamin C on HO-1
Several studies suggested a potential role of ferritin in protecting cells from oxidative damage through the sequestration of intracellular free iron10,11,12. Among enzymes promoting ferritin expression, the stress-inducible HO-1 has been implicated in promoting cell survival during cell exposure to oxidative insults29,30.
Since the FMD/STS downregulates the FTH protein expression level, we investigated whether HO-1 is implicated in FTH regulation in response to STS and vitamin C treatment. In one recent study from our group, the FMD sensitized breast cancer cells to chemotherapy in part by downregulating HO-1, further supporting a possible role of this stress-inducible protein in mediating FMD beneficial effects31.
To test our hypothesis, we evaluated the association between HO-1 expression and FTH induction. To this end, we treated HCT116 cells with the HO-1-activator hemin and verified that hemin increases both HO-1 and FTH protein levels under CTR and STS growing conditions (Supplementary Fig. 5).
Furthermore, we found that treatment with vitamin C significantly upregulated HO-1, while FMD/STS reversed this effect both in vitro and in vivo in KRAS-mutant cancer cells (Fig. 4a and Supplementary Fig. 6a).
In KRAS-wild-type cancer cells HO-1 levels were not altered upon vitamin C administration under CTR condition, and the combination of vitamin C and STS did not downregulate but instead induced HO-1 protein expression level (Fig. 4b, Supplementary Fig. 6b).
Fig. 4: HO-1 modulation and iron-bound transferrin are the key players in FMD-dependent sensitization to Vitamin C.
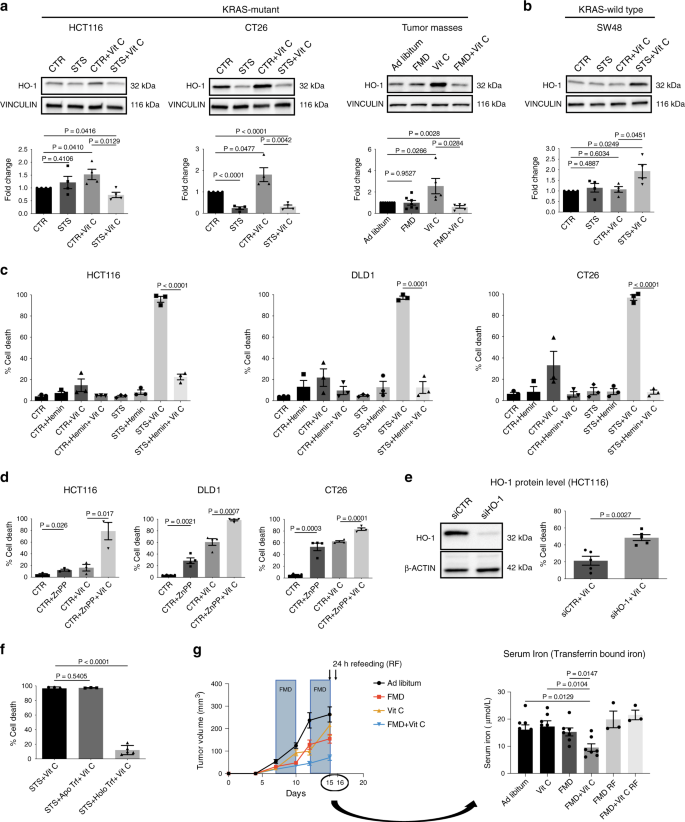
Collectively, these findings indicate that the differential regulation of HO-1 and the resulting effect on ferritin/iron pathway mediate the FMD-dependent sensitization to vitamin C selectively in a KRAS-mutant background.
The ability of the FMD to prevent vitamin C-induced HO-1 upregulation in KRAS-mutant cancer cells prompted us to investigate whether HO-1 levels affect the sensitivity of tumor cells to the combination treatment. Consistent with this hypothesis, the HO-1 activator hemin protected human and murine KRAS-mutant CRC cells from vitamin C-induced cell death, alone or in combination with STS (Fig. 4c).
On the other hand, the HO-1 inhibitor zinc protoporphyrin (ZnPP) made KRAS-mutant CRC cells more susceptible to vitamin C in nutrient-rich condition (CTR) (Fig. 4d). Consistent with these findings, HO-1 knockdown in HCT116 cells also increased cancer cell death upon vitamin C exposure in CTR condition (Fig. 4e). Thus, overall these results support the role of HO-1 in regulating KRAS-mutant cancer cell sensitivity to vitamin C.
To characterize how FMD/STS affects HO-1 levels and modulate cancer cell sensitivity to vitamin C, we analysed the effect of glucose or serum deprivation. Interestingly we found that glucose, serum growth factors and amino acids were not responsible for the STS-dependent enhancement of vitamin C toxicity (Supplementary Fig. 7a).
Addition of holo-transferrin (iron-bound form) but not apo-transferrin (iron-free form) reversed STS + vitamin C-mediated toxicity and HO-1/FTH axis downregulation (Fig. 4f; Supplementary Fig. 7b). These results are consistent with the concept that iron levels in the serum are important in mediating the STS effect. In fact, in agreement with our in vitro findings, in vivo FMD + vitamin C reduced blood levels of transferrin bound iron (Fig. 4g). Our data further support the role of iron as a key factor in the serum whose reduction was responsible for the synergism of FMD and vitamin C.
FMD and vitamin C potentiate oxaliplatin cytotoxic effects
A number of studies described the tolerability and potential efficacy of high-dose vitamin C as an adjuvant treatment during chemotherapy2,3,32,33. In addition, our group has recently shown the effectiveness of fasting or FMD cycles in combination with chemotherapy to reduce tumor growth in a wide range of cancer types compared with standard chemotherapy alone17,31.
Drawing from our previous data, we investigated whether FMD + vitamin C would sensitize KRAS-mutated cancer cells to the pro-oxidant action of chemotherapy in vivo, possibly by increasing cellular oxidative stress.
We chose oxaliplatin because it is one of the most effective cytotoxic compounds used in the adjuvant and advanced setting of CRC treatment34. Of note, the FMD + vitamin C combination was as effective as oxaliplatin + FMD or oxaliplatin + vitamin C, supporting the powerful action of these non-toxic combinations in halting tumor growth (Fig. 5a).
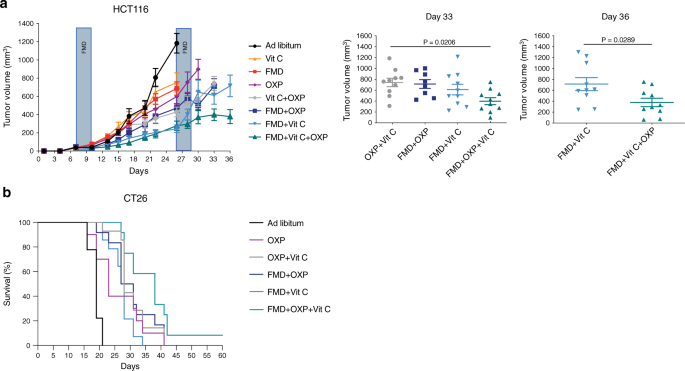
NSG and BALB/c mice were subcutaneously injected with HCT116 cells and CT26 cells, respectively. Mice were fed ad libitum or subjected to FMD cycles, and treated with or without vitamin C or oxaliplatin (10 mg/kg). a HCT116 tumor progression (left) and volume at day 33 and 36 (right), respectively (n = 10 in Ad libitum, FMD, FMD + Vit C, Vit C + OXP, FMD + Vit C + OXP, n = 8 in FMD + OXP, n = 9 in OXP, n = 11 in Vit C). P values were determined by One-way ANOVA with Tukey’s post analysis (day 33) and two-sided unpaired t-test (day 36). Data are represented as mean ± SEM. b BALB/c with CT26 survival curves (n = 9 in Ad libitum, n = 10 in OXP, n = 12 in FMD + OXP and FMD + OXP + Vit C, n = 14 in OXP + Vit C and FMD + Vit C). P values were determined by Log-rank (Mantel-Cox) test (Ad libitum vs OXP, p = 0.0040; OXP vs FMD + Vit C, p = 0.7177; OXP vs FMD + OXP + VitC, p = 0.0114; FMD + OXP vs FMD + OXP + Vit C, p = 0.0488; OXP + Vit C vs FMD + OXP + Vit C, p = 0.0345; FMD + OXP + Vit C vs FMD + Vit C, p = 0.0003).
Moreover, triple treatment (FMD + vitamin C + chemotherapy) was the most active therapeutic intervention in delaying tumor progression in a mouse xenograft and in extending survival in a syngeneic model (Fig. 5a, b and Supplementary Fig. 8).
These results indicate that chemotherapy can further potentiate the effects of FMD + vitamin C against KRAS mutated cancers.
More information: Maira Di Tano et al, Synergistic effect of fasting-mimicking diet and vitamin C against KRAS mutated cancers, Nature Communications (2020). DOI: 10.1038/s41467-020-16243-3
References
1. Hoffer, L. J. et al. Phase I clinical trial of i.v. ascorbic acid in advanced malignancy. Ann. Oncol. 19, 1969–1974 (2008).
2.Ma, Y. et al. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci. Transl. Med. 6, 222ra18 (2014).
3. Schoenfeld, J. D. et al. O2⋅- and H2O2-mediated disruption of Fe metabolism causes the differential susceptibility of NSCLC and GBM cancer cells to pharmacological ascorbate. Cancer Cell 32, 268 (2017).
4. Yun, J. et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 350, 1391–1396 (2015).
5.Aguilera, O. et al. Vitamin C uncouples the Warburg metabolic switch in KRAS mutant colon cancer. Oncotarget 7, 47954–47965 (2016).
6. Chen, Q. et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc. Natl Acad. Sci. USA 104, 8749–8754 (2007).
7.Chen, Q. et al. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc. Natl Acad. Sci. USA 105, 11105–11109 (2008).
8.Du, J. et al. Mechanisms of ascorbate-induced cytotoxicity in pancreatic cancer. Clin. Cancer Res. 16, 509–520 (2010).
9.Moser, J. C. et al. Pharmacological ascorbate and ionizing radiation (IR) increase labile iron in pancreatic cancer. Redox Biol. 2, 22–27 (2013).
10.Torti, S. V. & Torti, F. M. Iron and cancer: more ore to be mined. Nat. Rev. Cancer 13, 342–355 (2013).
11.Kakhlon, O., Gruenbaum, Y. & Cabantchik, Z. I. Ferritin expression modulates cell cycle dynamics and cell responsiveness to H-ras-induced growth via expansion of the labile iron pool. Biochem. J. 363, 431–436 (2002).
12.Yang, W. S. & Stockwell, B. R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 15, 234–245 (2008).
13.Ibrahim, W. H., Habib, H. M., Kamal, H., Clair, D. K. S. & Chow, C. K. Mitochondrial superoxide mediates labile iron level: evidence from Mn-SOD-transgenic mice and heterozygous knockout mice and isolated rat liver mitochondria. Free Radic. Biol. Med. 65, 143–149 (2013).
14.Caltagirone, A., Weiss, G. & Pantopoulos, K. Modulation of cellular iron metabolism by hydrogen peroxide. Effects of H2O2 on the expression and function of iron-responsive element-containing mRNAs in B6 fibroblasts. J. Biol. Chem. 276, 19738–19745 (2001).
15.Otterbein, L. E., Soares, M. P., Yamashita, K. & Bach, F. H. Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol. 24, 449–455 (2003).
16.Stephen, A. G., Esposito, D., Bagni, R. K. & McCormick, F. Dragging ras back in the ring. Cancer Cell 25, 272–281 (2014).
17.Lee, C. et al. Fasting cycles retard growth of tumors and sensitize a range of cancer cell types to chemotherapy. Sci. Transl. Med 4, 124ra27 (2012).
18.Raffaghello, L. et al. Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc. Natl Acad. Sci. USA 105, 8215–8220 (2008).
19.Lee, C. et al. Reduced levels of IGF-I mediate differential protection of normal and cancer cells in response to fasting and improve chemotherapeutic index. Cancer Res. 70, 1564–1572 (2010).
20.Lee, C. & Longo, V. D. Fasting vs dietary restriction in cellular protection and cancer treatment: from model organisms to patients. Oncogene 30, 3305–3316 (2011).
21.Longo, V. D. & Mattson, M. P. Fasting: molecular mechanisms and clinical applications. Cell Metab. 19, 181–192 (2014).
22.Di Biase, S. et al. Fasting regulates EGR1 and protects from glucose- and dexamethasone-dependent sensitization to chemotherapy. PLoS Biol. 15, e1002603 (2017).
23.Brandhorst, S. et al. A periodic diet that mimics fasting promotes multi-system regeneration, enhanced cognitive performance, and healthspan. Cell Metab. 22, 86–99 (2015).
24.Wei, M. et al. Fasting-mimicking diet and markers/risk factors for aging, diabetes. Cancer, Cardiovascular Dis. Sci. Transl. Med. 15, 9 (2017).
25.Bianchi, G. et al. Fasting induces anti-Warburg effect that increases respiration but reduces ATP-synthesis to promote apoptosis in colon cancer models. Oncotarget 6, 11806–11819 (2015).
26.Reczek, C. R. & Chandel, N. S. The two faces of reactive oxygen species in cancer. Annu. Rev. Cancer Biol. 1, 79–98 (2017).
27.Weinberg, F. et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl Acad. Sci. USA 107, 8788–8793 (2010).
28.Ogrunc, M. et al. Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ. 21, 998–112 (2014).
29.Ferris, C. D. et al. Haem oxygenase-1 prevents cell death by regulating cellular iron. Nat. Cell Biol. 1, 152–157 (1999).
30.Gonzales, S., Erario, M. A. & Tomaro, M. L. Heme oxygenase-1 induction and dependent increase in ferritin. Dev. Neurosci. 24, 161–168 (2002).
31.Di Biase, S. et al. Fasting-mimicking diet reduces HO-1 to promote T cell-mediated tumor cytotoxicity. Cancer Cell 30, 136–146 (2016).
32.Monti, D. A. et al. Phase I evaluation of intravenous ascorbic acid in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. PLoS ONE 7, e29794 (2012).
33.Welsh, J. L. et al. Pharmacological ascorbate with gemcitabine for the control of metastatic and node-positive pancreatic cancer (PACMAN): results from a phase I clinical trial. Cancer Chemother. Pharmacol. 71, 765–775 (2013).
34.Alcindor, T. & Beauger, N. Oxaliplatin: a review in the era of molecularly targeted therapy. Curr. Oncol. 18, 18–25 (2011).
35.Muliaditan, T. et al. Repurposing tin mesoporphyrin as an immune checkpoint inhibitor shows therapeutic efficacy in preclinical models of cancer. Clin. Cancer Res. 24, 1617–1628 (2018).
36.Was, H., Dulak, J. & Jozkowicz, A. Heme oxygenase-1 in tumor biology and therapy. Curr. Drug Targets 11, 1551–1570 (2010).
37.Matsuo, T. et al. Pathological significance and prognostic implications of heme oxygenase 1 expression in non-muscle-invasive bladder cancer: Correlation with cell proliferation, angiogenesis, lymphangiogenesis and expression of VEGFs and COX-2. Oncol. Lett. 13, 275–280 (2017).
38.Deng, Y. et al. The Nrf2/HO-1 axis can be a prognostic factor in clear cell renal cell carcinoma. Cancer Manag. Res. 7, 1221–1230 (2019).
39.Sun, W. et al. TSVdb: a web-tool for TCGA splicing variants analysis. BMC Genomics. 19, 405 (2018).



{kind=link}