










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



All viruses mutate as they make copies of themselves to spread and thrive. SARS-CoV-2, the virus the causes COVID-19, is proving to be no different. There are currently more than 4,000 variants of COVID-19, which has already killed more than 2.7 million people worldwide during the pandemic.
The UK variant, also known as B.1.1.7, was first detected in September 2020, and is now causing 98 percent of all COVID-19 cases in the United Kingdom. And it appears to be gaining a firm grip in about 100 other countries it has spread to in the past several months, including France, Denmark, and the United States.
The World Health Organization says B.1.1.7 is one of several variants of concern along with others that have emerged in South Africa and Brazil.
“The UK, South Africa, and Brazil variants are more contagious and escape immunity easier than the original virus,” said Victor Padilla-Sanchez, a research scientist at The Catholic University of America.
“We need to understand why they are more infectious and, in many cases, more deadly.”
All three variants have undergone changes to their spike protein – the part of the virus which attaches to human cells. As a result, they are better at infecting cells and spreading.
In a research paper published in January 2021 in Research Ideas and Outcomes, Padilla-Sanchez discusses the UK and South African variants in detail. He presents a computational analysis of the structure of the spike glycoprotein bound to the ACE2 receptor where the mutations have been introduced.
His paper outlines the reason why these variants bind better to human cells.
“I’ve been analyzing a recently published structure of the SARS-CoV-2 spike bound to the ACE2 receptor and found why the new variants are more transmissible,” he said.
“These findings have been obtained using UC San Francisco Chimera software and molecular dynamics simulations using the Frontera supercomputer of the Texas Advanced Computing Center (TACC).”
Padilla-Sanchez found that the UK variant has many mutations in the spike glycoprotein, but most important is one mutation, N501Y, in the receptor binding domain that interacts with the ACE2 receptor.
“This N501Y mutation provides a much higher efficiency of binding, which in turn makes the virus more infectious. This variant is replacing the previous virus In the United Kingdom and is spreading in many other places in the world,” he said.
The South Africa variant emerged in October 2020, and has more important changes in the spike protein, making it more dangerous than the UK variant. It involves a key mutation—called E484K—that helps the virus evade antibodies and parts of the immune system that can fight coronavirus based on experience from prior infection or a vaccine.
Since the variant escapes immunity the body will not be able to fight the virus. “We’re starting to see the South Africa variant here in the U.S.,” he said.
Padilla-Sanchez performed structural analysis, which studied the virus’s crystal structure; and molecular dynamics to obtain these findings.
“The main computational challenge while doing this research was to find a computer powerful enough to do the molecular dynamics task, which generates very big files, and requires a great amount of memory. This research would not have been possible without the Frontera supercomputer,” Padilla-Sanchez said.
According to Padilla-Sanchez, the current vaccines will not necessarily treat the variants. “The variants will require their own specific vaccines. We’ll need as many vaccines for variants that appear.”
Going forward, Padilla-Sanchez will continue to research the changes taking place with SARS-CoV-2.
“This was a very fast project—the computational study lasted one month,” he said. “There are many other labs doing wet lab experiments, but there aren’t many computational studies. That’s why I decided to do this important work now.”
Two new severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) lineages carrying the amino acid substitution N501Y in the receptor-binding domain (RBD) of the spike protein have spread rapidly in the United Kingdom (UK) during late autumn 2020.
Assessing the public health threat of these lineages (e.g. the potential for them to increase herd immunity thresholds if they displace other circulating SARS-CoV-2 strains) requires quantification of their comparative transmissibility. Here we adopted our previous epidemiological framework for relative fitness inference of co-circulating pathogen strains, which has been applied on influenza viruses [1] and SARS-CoV-2 D614G strains [2], to characterise the comparative transmissibility of the 501Y lineages.
Table.Genetic changes that characterise 501Y Variant 1 and Variant 2a and occurred in the genetic branches preceding their lineages

a 501Y Variant 2 was also named B.1.1.7 by COVID-19 Genomics Consortium UK (CoG-UK) [3], 20B/501Y.V1 by Nextstrain (https://nextstrain.org/) and VOC-202012/01 by Public Health England [4] and assigned clade GR as per the GISAID initiative on common clade nomenclature system (www.gisaid.org)*.
The most concerning mutation is indicated in bold.
The most concerning mutation is N501Y, which co-occurs with several mutations of potential biological importance, including P681H and deletion of the amino acid at the 69th and 70th residues (Δ69/Δ70) on the spike protein (Supplementary Table S1). Structural biological studies of the SARS-CoV-2 RBD offer insights proposing that 501Y may increase human angiotensin-converting enzyme 2 (ACE2) binding [6,7] and that the open conformation of the 501Y spike protein [8] is associated with more efficient viral entry and infection. Epidemiologically, however, there has been limited assessment to date investigating whether any of these mutations may have affected transmissibility [9].
We downloaded the multiple sequence alignment of complete (or nearly complete) genomes of SARS-CoV-2 from the GISAID database initially on 14 December. To include more sequences for the study, we extended our search for 501N and 501Y sequences in the GISAID dataset downloaded on 19 December, including both the complete genomes and partial ones covering spike genes.
We extracted all viral genomes carrying 501Y in the translated spike protein and analysed them with other closely related virus strains (identified through basic local alignment search tool (BLAST) search) in the global phylogeny (Supplementary Table S2). The resultant phylogeny built with the maximum likelihood method and generalised time-reversible (GTR) substitution model using FastTree version 2.1 [10] is shown in Figure 1. It indicates that the recent 501Y strains in the UK, since August/September 2020, emerged from the 20B clade (Nextstrain nomenclature) and formed two lineages. Both lineages have clear geographical separation in Wales vs England.
The first 501Y lineage (501Y Variant 1) appeared in Wales in early September and persisted through November. The second 501Y lineage (501Y Variant 2, also named B.1.1.7, 20B/501Y.V1 and VOC-202012/01 and assigned clade GR*) appeared in England in late September and largely expanded to become the predominant lineage in the region since late November. Globally, two other lineages with 501Y (without Δ69/Δ70) have been detected in Australia and South Africa, circulating from June to July and October to November 2020, respectively.
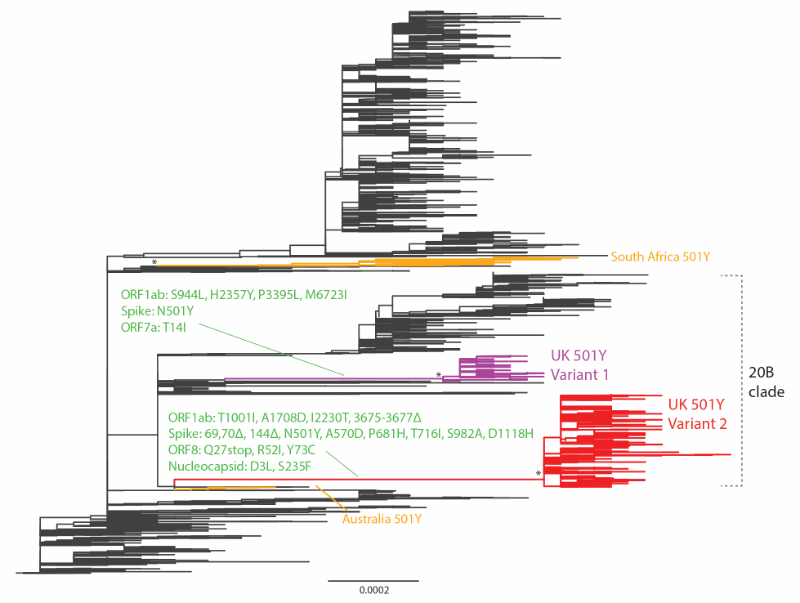
Nt: nucleotide; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2; UK: United Kingdom; US: United States.
The maximum likelihood tree was built from 7,003 genome nt sequences of SARS-CoV-2. The UK 501Y Variant 1 and 2 lineages are coloured in purple and red, respectively. The South African and Australian 501Y lineages are in orange. Amino acid changes at the preceding branches of UK 501Y Variant 1 and 2 are indicated in green. Some 501Y variants in sporadic emergence (including those in Spain and the US, etc.) without establishment of a lineage of more than 10 sequences are not shown. The asterisks refer to the > 0.98 topology supports (Shimodaira-Hasegawa test) for the monophyletic grouping of the 501Y lineages. Branch scale is shown at the bottom of the tree in the unit of substitutions per site.
We assumed that the N501Y mutation and Δ69/Δ70 deletions characterise the three strains (501N, 501Y Variant 1 and 501Y Variant 2), but their differential transmissibility (if any) might be attributable to the combination of N501Y and other mutations acquired in the emergence of 501Y Variant 1 and 2 lineages (Table and Supplementary Table S1). For conciseness, we used N, Y1 and Y2 to denote the three strains. We defined the comparative transmissibility of any two strains as the ratio of their basic reproductive numbers (R0) (R0) . That is, the comparative transmissibility of strains Y1 and Y2 with respect to strain N was σY1=RY10/RN0σY1=R0Y1/R0N and σY2=RY20/RN0σY2=R0Y2/R0N , respectively.
We extended the previous competition transmission model of two viruses [1,2] and applied the fitness inference framework to the sequence data collected from the UK between 22 September and 16 November 2020, during the co-circulation period of the three strains (see Supplementary Material for details).
The inference framework incorporates both incidence and genotype frequency data that reflect the local comparative transmissibility of co-circulating strains. Using confirmed deaths (adjusted for the delay between symptom onset and death [11]) as the proxy for the coronavirus disease (COVID-19) epidemic curve [12], we estimated that σY1σY1 was 1.10 (95% credible interval (CrI): 1.06–1.13) and σY2σY2 was 1.75 (95% CrI: 1.70–1.80).
That is, the R0R0 of the 501Y Variant 1 and Variant 2 was 10% (95% CrI: 6–13%) and 75% (95% CrI: 70–80%) higher, respectively, than that of the 501N strain.
The fitted model was largely congruent with the observed proportions of the three strains over time, except during 13–19 October and 3–9 November, for 501Y Variant 1 (Figure 2-3). Since 501Y Variant 1 mainly co-circulated with 501N in Wales, we also performed a separate analysis using sequence data from Wales only.
We estimated σY1σY1 was 1.14 (95% CrI: 1.11–1.19) but were not able to estimate σY2σY2 because there were only two 501Y Variant 2 sequences sampled before 30 November from Wales in our dataset.
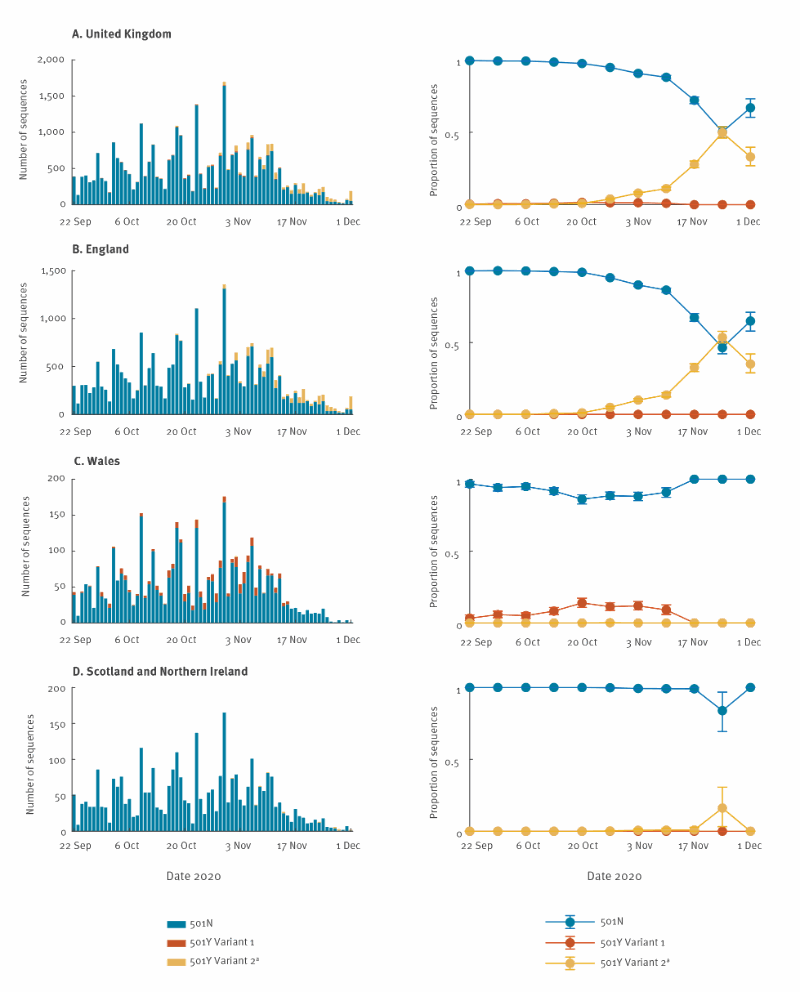
CI: confidence interval; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2; UK: United Kingdom.
a 501Y Variant 2 was also named B.1.1.7 by COVID-19 Genomics Consortium UK (CoG-UK) [3], 20B/501Y.V1 by Nextstrain (https://nextstrain.org/) and VOC-202012/01 by Public Health England [4] and assigned clade GR as per the GISAID initiative on common clade nomenclature system (www.gisaid.org)*.
The circles and error bars indicate the observed proportion, with 95% multinomial CIs among sequence data.
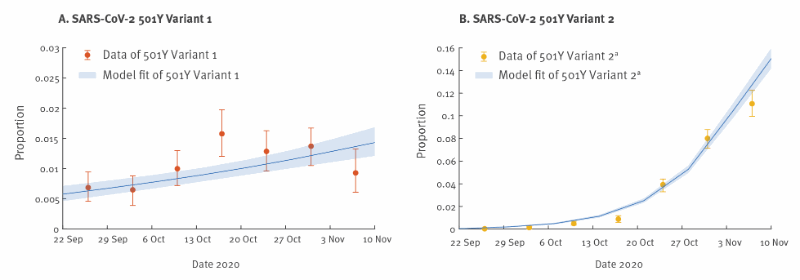
CI: confidence interval; COVID-19: coronavirus disease; CrL: Credible interval; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2; UK: United Kingdom.
a 501Y Variant 2 was also named B.1.1.7 by COVID-19 Genomics Consortium UK (CoG-UK) [3], 20B/501Y.V1 by Nextstrain (https://nextstrain.org/) and VOC-202012/01 by Public Health England [4] and assigned clade GR as per the GISAID initiative on common clade nomenclature system (www.gisaid.org)*.
The time series of confirmed COVID-19 deaths in the UK was used in the estimation of comparative transmissibility. The circles and error bars indicate the observed proportion, with 95% multinomial CIs among sequence data. The blue lines and shades indicate the posterior mean and 95% CrI of the estimated proportions of 501Y lineages.
Sensitivity analyses to assess the possible impact of generation times on findings
We conducted a sensitivity analysis to assess the possibility that the transmission advantages of 501Y lineages were due to shorter generation time [2]. Assuming the same R0 for the three strains, we estimated the mean generation time of 501Y Variant 2 was 44% (95% CrI: 39–47%) shorter than that of 501N, but the inference failed to converge to generate estimates for 501Y Variant 1. Moreover, this fitted model had significantly higher Akaike information criterion (AIC) than our base case model, hence favouring our base case conclusion that the transmission advantage of 501Y Variant 2 was due to higher R0 but not shorter generation time.
reference link:https://www.eurosurveillance.org/content/10.2807/1560-7917.ES.2020.26.1.2002106
More information: Victor Padilla-Sanchez. SARS-CoV-2 Structural Analysis of Receptor Binding Domain New Variants from United Kingdom and South Africa, Research Ideas and Outcomes (2021). DOI: 10.3897/rio.7.e62936



{kind=link}
[…] COVID-19: UK – South Africa coronavirus variants escape immunity by N501Y… […]