











 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Researchers from Critical Analytics for Manufacturing Personalized-Medicine (CAMP), an Interdisciplinary Research Group (IRG) at the Singapore-MIT Alliance for Research and Technology (SMART), MIT’s research enterprise in Singapore, have developed a new method for rapid and accurate detection of viral nucleic acids – a breakthrough that can be easily adapted to detect different DNA/RNA targets in viruses like the coronavirus.
The pandemic has highlighted the importance of rapid diagnostics and improved methods to detect viruses, especially as the world seeks to be prepared for future pandemics or the next dangerous pathogen.
Particularly, the biomanufacturing industry, with the unique challenges of using cells as cell therapy products, is looking for innovations in rapid methods to detect virus contamination as part of their quality control processes and in release testing.
A more accurate version is the digital PCR method that allows absolute quantification – meaning it reveals the copy number of viruses in a sample – can allow for setting clear thresholds of virus contamination, and is not susceptible to potential fluctuations of reference gene required by standard qPCR methods.
However, digital PCR demands a relatively long reaction time of around four hours. Another drawback of all current PCR-based methods is that they need expensive equipment for precise temperature control and cycling.
The new methodological development by CAMP – the RApid DIgital Crispr Approach (RADICA) – allows absolute quantification of viral nucleic acids in 40-60 minutes in an isothermal manner in a water bath, a prototypical and inexpensive laboratory equipment.
The team’s research is explained in a paper titled “Digital CRISPR-based method for the rapid detection and absolute quantification of nucleic acids” published recently in the prestigious journal Biomaterials.
The RADICA method has been tested on SARS-CoV-2 synthetic DNA/RNA as well as the Epstein-Barr virus in cultured B cells and patient serum. The researchers say the method can be adapted to detect other kinds of viruses, and in other types of samples such as saliva and cell culture media. RADICA is also able to distinguish the virus from their close relatives.
“This is the first reported method of detecting nucleic acids to utilize the sensitivity of isothermal amplification and specificity of CRISPR based detection in a digital format – allowing rapid and specific amplification of DNA without the time consuming and costly need for thermal cycling,” says Dr. Xiaolin Wu, Postdoctoral Associate at SMART CAMP. “RADICA offers four times faster absolute quantification compared to conventional digital PCR methods.”
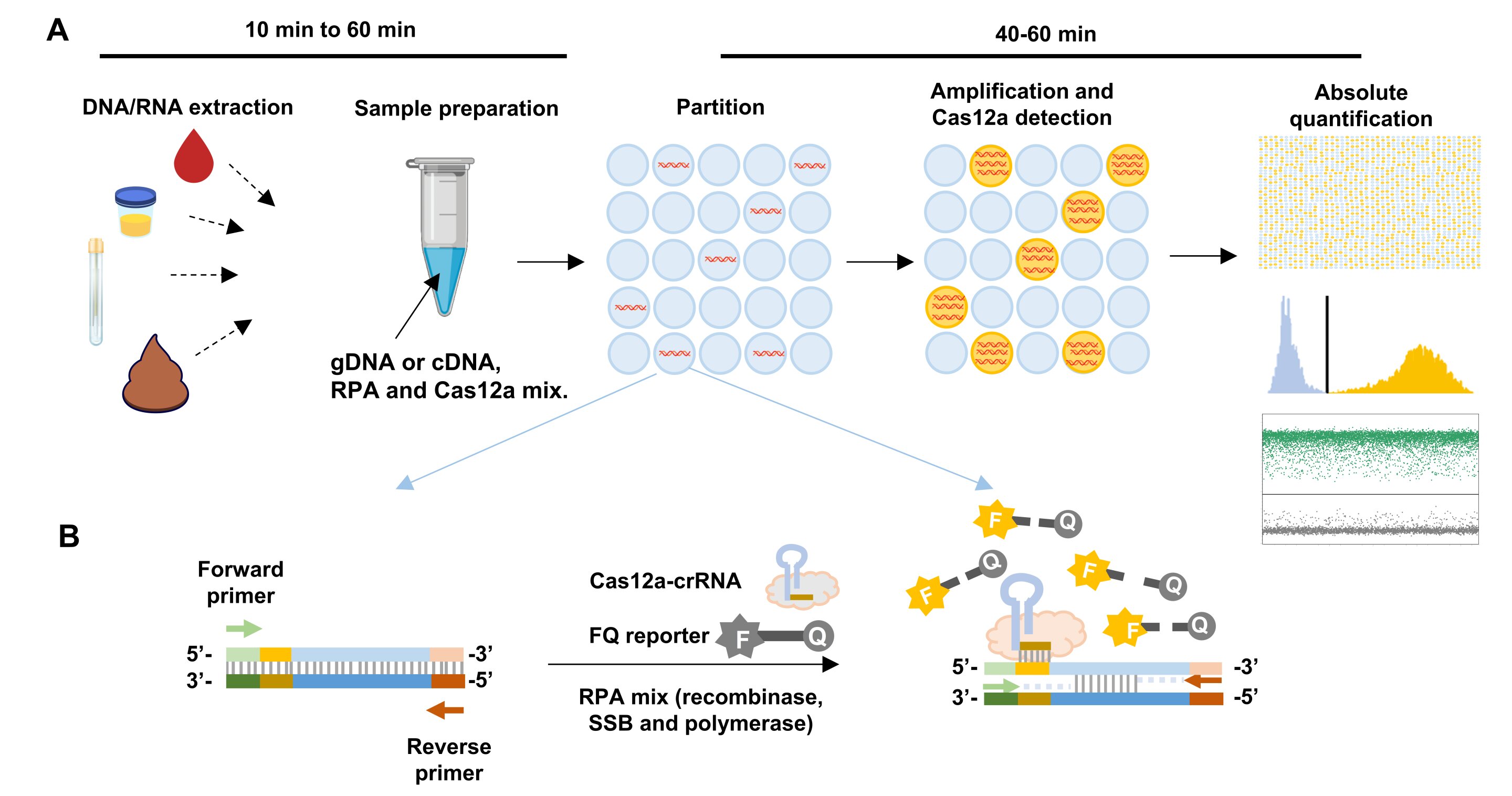
The team uses extracted DNA/RNA of the sample and divides a 15 µL reaction into thousands of independent partitions. In each partition, the DNA/RNA is amplified and identified by Cas12a protein, an enzyme that can turn the target signal into a fluorescent signal. This allows absolute quantification to be achieved by counting the number of partitions that have the target DNA/RNA and are lit up.
“The last year has shown us the importance of detecting viruses quickly and accurately, and RADICA can help fill existing gaps in this area,” says National University of Singapore Professor Hanry Yu, co-corresponding author and Co-Lead Principal Investigator at SMART CAMP. “Cell therapy products have a very short shelf life and patients are usually in need of treatment urgently. Current sterility tests need around 14 days, which is too slow for clinical needs but RADICA shortens the process into hours.”
Professor Tim Lu, who is co-corresponding author, CAMP Principal Investigator and Associate Professor of Biological Engineering and Electrical Engineering and Computer Science at Massachusetts Institute of Technology, said the team’s method is faster, cheaper and more efficient than what is used today and its digital format makes it more tolerant to contamination or inhibitors that may be present in biological samples – often the case with cell therapy products.
Professor Lu adds that on top of detecting the presence of a target virus, RADICA also identifies how many viruses there are in the sample which can help doctors and researchers in deciding the course of treatment, as well as production and inventory management of cell therapy products.
While the researchers at CAMP developed RADICA for monitoring cell therapy manufacturing processes and biosafety release testing of cell therapy products, Dr. Wu says the method can also be used to detect DNA/RNA targets of different viruses and adapted to devices commonly found in hospitals and service laboratories—providing a potential new way to tackle pandemics.
Design of RADICA
Commercial chips for sample partitioning and matched fluorescence reader for endpoint detection were used in RADICA23. In this system, each CRISPR-based reaction mix is sub-divided into 10,000 partitions on the chip, resulting in an average partition volume of 1.336 nL. We first optimized the bulk CRISPR reaction to achieve a one-copy-per-1.336 nL partition detection sensitivity on the chip. This is equivalent to femtomolar detection sensitivity in a bulk reaction.
We selected the Cas12a homolog from Lachnospiraceae bacterium ND2006 (LbCas12a) as it showed the highest signal-to-noise ratio relative to other Cas12a homologs from a previous study17. To test if the RADICA could detect viral DNA with femtomolar sensitivity without pre-amplification, serially-diluted dsDNA (double stranded DNA) was incubated with LbCas12a together with its CRISPR RNA (crRNA) and a reporter (quenched fluorescent DNA). The sensitivity of detection using the CRISPR-based method without pre-amplification in a bulk reaction was found to be 10 pM (Supplementary Fig. 1), which did not meet the femtomolar sensitivity requirement of RADICA.
To increase the sensitivity of detection of the CRISPR-based method, an isothermal amplification step was used. RPA was chosen for the isothermal amplification step because its reaction temperature (25°C to 42°C) is compatible with that of Cas12a (25°C to 48°C). This allowed for a one-step digital RPA-CRISPR absolute quantification method that eliminates multiple manipulations inherent in two-step CRISPR-based detection methods such as SHERLOCK and DETECTR14, 15.
To avoid Cas12a-mediated cleavage of the target molecule before amplification, we designed the crRNA to target single-stranded DNA (ssDNA) that is generated only after amplification of the target molecule (Fig. 1b)22. Another advantage of this method is the ease of designing ssDNA-targeting crRNA over traditional dsDNA-targeting crRNA, because the nuclease activity of Cas12a on ssDNA has been reported to be independent of the presence of protospacer adjacent motif (PAM)24.
The RADICA developed in this study is illustrated in Fig. 1a. Extracted DNA samples are loaded onto on the chip by capillary action, and the reaction is partitioned into 10,000 compartments, resulting in zero or one target molecule in each compartment. To prevent spontaneous target amplification by RPA at room temperature25, the RPA-CRISPR reaction was prepared without the addition of Mg2+, which is required for the polymerase activity. All reactions were prepared on ice and samples were loaded within one minute to prevent premature target amplification.
The partitioned reactions were incubated in isothermal water baths, heat blocks, or warm rooms.In each compartment containing the target molecule (Fig. 1b), RPA initiates from one DNA strand and subsequently exposes the crRNA-targeted ssDNA region on the other strand. As the amplification proceeds, Cas12a cleaves the positive ssDNA strand, triggering its collateral cleavage activity, which in turn cleaves the proximal quenched fluorescent probe (ssDNA-FQ reporter) to generate a fluorescence signal.
At the same time, ongoing amplification of the other DNA strand exponentially amplifies the target DNA, triggering more Cas12a activation and increasing the fluorescence readout. The proportion of positive-to-negative compartments is analyzed based on the endpoint fluorescence measurement, and the copy number of the target nucleic acid is calculated based on the Poisson distribution, allowing for absolute quantification of the sample (Fig. 1a). Concurrent detection of compartments in each tube by the Clarity™ Reader shortens detection time to within minutes.
RADICA optimization
To validate and optimize the RADICA, G-block DNA or plasmids containing the SARS-CoV-2 N (nucleoprotein) gene region were used and primers and crRNAs specific for the SARS-CoV-2 N gene were designed accordingly based on previous studies22. The target regions overlap with those of the China CDC assay (N gene region) with some modification to meet the primer and crRNA design (Supplementary Table 1).
To optimize the Cas12a-mediated reaction, a bulk reaction using 0.1 nM and 1 nM dsDNA as a target was performed with a range of Cas12a/crRNA concentrations. We found that in the presence of a constant amount of target DNA and probe, comparable fluorescence signal intensities were detected between 50 nM to 250 nM Cas12a-crRNA concentrations, suggesting that changing the Cas12a/crRNA concentration did not influence the reaction (Supplementary Fig. 2).
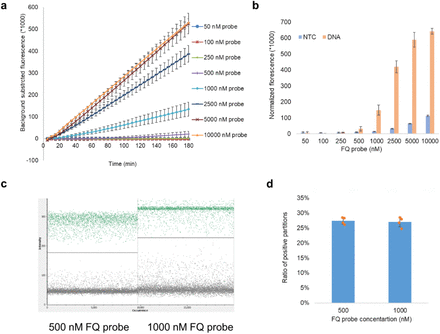
Optimization of FQ probe concentration in RADICA.
a, b, Cas12a reaction in bulk reactions with different FQ probe concentrations in the presence or absence of a constant concentration (0.1 nM) of target DNA. a, Time course reaction of Cas12a with FQ probes at concentrations ranging from 50 nM to 10,000 nM. X-axis indicates the reaction time; y-axis indicates the background-subtracted fluorescence signal. b, Fluorescence signal of DNA and non-template control obtained with FQ probes at concentrations ranging from 50 nM to 10,000 nM. c, d, RADICA reaction with the same concentrations of target DNA but different probe concentrations. c, Fluorescence intensity of the negative partitions (background noise) and positive partitions (positive signals) on the chip obtained with FQ probes at concentrations of 500 or 1000 nM. d, Histogram showing ratios of positive partitions on the chip with FQ probes, at concentrations of 500 or 1000 nM, in the presence of target DNA (4 replicates for each FQ probe concentration).
Since the quenched fluorescent probe is another key component that influences the reaction, we optimized the FQ assay by incubating increasing amounts of FQ probes with constant concentrations of Cas12a-crRNA and target DNA. As expected, CRISPR-mediated fluorescence signal intensities increased with increasing amounts of FQ probes (from 250 nM to 5 µM), although higher probe concentrations also resulted in higher background noise (Fig. 2a,b).
At FQ probe concentrations above 5 µM, the signal-to-noise ratio could not be further enhanced (Fig. 2b). To ensure that the fluorescence signal generated on the partitioned chip was within the reader’s detection range, different FQ probe concentrations were tested in independent digital CRISPR reactions in the presence of the target DNA and the fluorescence measured on the digital PCR fluorescence reader. We found that in the presence of the same target DNA, the proportions of positive partitions were comparable regardless of the FQ probe concentration used (Fig. 2d).
However, only the background noise and positive signals generated in the reaction with 500 nM FQ probe concentration were within the reader’s detection range, while the reactions containing 1000 nM FQ probe concentration yielded higher background noises, which are difficult to separate from positive signals (Fig. 2c). We therefore used 500 nM FQ probe concentrations to achieve high signal-to-noise ratios for subsequent experiments.
An additional optimization step involved developing a one-pot reaction that combines the RPA and Cas12a reactions. We performed the bulk reaction at 25°C, 37°C and 42°C, which are temperatures within the reaction temperature ranges of RPA (25°C to 42°C) and Cas12a (25°C to 48°C). First, we tested the reaction using serial dilutions of plasmid DNA at 25°C and 42°C.
The reaction proceeded at both of these reaction temperatures, with a limit of detection of about 9.4 copies/µL. However, at 25°C, the reaction proceeded significantly more slowly with lower positive signals and a higher background than the reaction performed at a 42°C (Supplementary Fig. 3).
Next, we assessed the effect of different temperatures (25°C, 37°C and 42°C) on reactions containing a constant amount of plasmid DNA (37.5 copies/µL). We found that higher temperatures accelerated the reaction (Supplementary Fig. 3c). Taken together, our results suggest that 42°C is the optimal temperature for the RPA-Cas12a reaction.
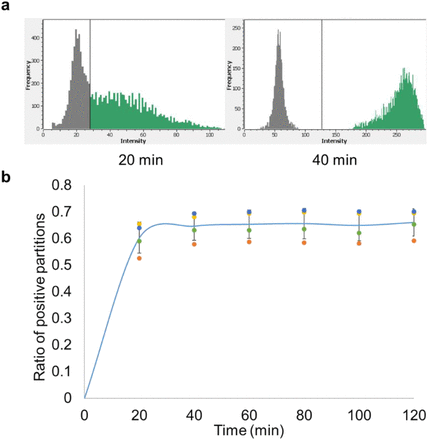
Time course reaction of RADICA.
a, Fluorescence intensity of the partitions on the chip at two time points. The x-axis represents fluorescence intensity while the y-axis represents the frequency of the partitions. The left peak (low fluorescence level; dark grey) on the fluorescence intensity histogram represents the negative partitions while the right peak (high fluorescence level; green) indicates the positive partitions. As the CRISPR reaction proceeds, the fluorescence levels of the positive partitions increase and the right peak shifts further to the right. b, The proportion of positive partitions at different time points of RADICA. Each DNA replicate is represented by a data point with a unique color. Starting at about 60 minutes, the fluorescence signal plateaus and the ratio of positive partitions
We next investigated whether the reaction time affected the precision of RADICA on plasmid DNA detection at 42°C. As shown in Fig. 3a, the reaction proceeded quickly with some fluorescence signal detected in several compartments at 20 min, but with a low signal-to-noise ratio at this time point.
As the reaction proceeded, two distinct peaks indicating the negative (left) and positive (right) partitions were detected at 40 min (Fig. 3a). Analysis of the ratio of positive partitions on the chip at the different time points revealed that the number of positive partitions reached a plateau after 60 min in all four replicates, suggesting that 60 min was the earliest end-point measurement (Fig. 3b). All subsequent experiments were therefore performed for 60 minutes.
reference link : https://www.medrxiv.org/content/10.1101/2020.11.03.20223602v3.full
More information: Xiaolin Wu et al, Digital CRISPR-based method for the rapid detection and absolute quantification of nucleic acids, Biomaterials (2021). DOI: 10.1016/j.biomaterials.2021.120876



{kind=link}