










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



B.1.1.519 Variant Researchers are warning that the Mexican variant B.1.1.519 which has been ravaging Mexico for a few months now not only causes an increased risk of hospitalization and disease severity but also increased the risk of mortality.
Further just like the Delta and Mu variants, it is spawning sub-variants that are of concern.
Two studies have already been published by Mexican researchers, one by detailing the emergence of the variant and the other describing its evolutionary landscape and its clinical impact in Mexico City.
https://link.springer.com/article/10.1007/s00705-021-05208-6
https://www.medrxiv.org/content/10.1101/2021.09.07.21262911v2
In the first study, the research team reported the identification of a potential variant of interest, harboring the mutations T478K, P681H, and T732A in the spike protein, within the newly named lineage B.1.1.519 that rapidly outcompeted the preexisting variants in Mexico and has been the dominant virus in the country during the first trimester of 2021.
The overall genome analysis of the viruses in the B.1.1.519 lineage showed the presence of 20 mutations in total, compared to the Wuhan-Hu-1 reference genome sequence (NCBI accession number MN908947). Eleven of these mutations are non-synonymous, and four of them are present in the spike protein. https://link.springer.com/article/10.1007/s00705-021-05208-6/tables/1
The B.1.1.519 variant is grouped in an independent clade derived from the clade 20B NextClade classification according to phylogenetic analysis. The B.1.1.519 variant is characterized by nine mutations, four of which are ORF1 substitutions and three spike substitutions.
Notably, a T478K mutation is present in the receptor binding domain (RBD), where mutations have been shown to reduce the activity of some monoclonal antibodies. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8053237/
The B.1.1.519 lineage, represented by the vast majority of the reported Mexican sequences, was first identified as a B.1.1.222 lineage. However, the presence of the mutations T478K, P681H, and T732A clearly differentiated it from this lineage, which does not contain these mutations, giving rise to the B.1.1.519 lineage.
An in silico analysis using different potent structures of related strains suggested that the position of the T478K mutation in the S protein is involved in antibody recognition and the receptor binding site.
In a deep mutational scanning of the SARS-CoV-2 receptor binding domain, the T478K mutation did not have a significant effect on folding or binding to human angiotensin-converting enzyme 2 (ACE2) . https://pubmed.ncbi.nlm.nih.gov/32841599/
It is however speculated that this mutation may be involved in immune evasion, particularly escape from antibody neutralization. https://pubmed.n cbi.nlm.nih.gov/33535027/
The P681H mutation is one of the mutations also found in the B.1.1.7 variant detected in the UK. It has been found to increase spike cleavage by furin-like proteases and some speculate that it might increase transmissibility.
The T732A mutation and the 69-70 deletion were also found in the in the spike protein. Both are believed to play roles in immune evasion.
In the second study, the research team found that the B.1.1.519 Mexican variant with the P681H, T478K, and T732A mutations was responsible for 90% of cases detected in February 2021 in Mexico City.
The team reported the effective reproduction number of B.1.1.519 and presents its geographical origin based on phylogenetic analysis. Also investigated was this variants evolution through haplotype analysis, and the most recent haplotypes were identified. The team also examined the clinical impact of patients infected with variant B.1.1.519 compared to individuals infected with non-B.1.1.519 SARS-CoV-2.
According to the study, the first patient infected with the B.1.1.519 variant in Mexico City was identified on November 3, 2020, only the second case recorded worldwide.
However within Mexico City, the variant frequency increased from 16% to 90% in February 2021 but decreased to 51% in May 2021.
It was found that of sequences generated from SARS-CoV-2 infections, variant B.1.1.519 represented 74.3% from November 2020 to May 2021 in Mexico City. The B.1.1.519 variant was detected in 31 countries, making up 55% of cases in Mexico.
In order to determine the transmissibility of variant B.1.1.519, the research team studied the effective reproduction number (Rt), which was defined as the average number of secondary cases per primary case at a given time within the year. It was observed that there was a sharp increase in the Rt for the B.1.1.519 variant, up to a value of 2.9 in the second week of December 2020, which corresponded with the increased detection. However, the Rt value began to stabilize in the following months, between 0.5 and 1.
Importantly the B.1.1.519 variant was the second most frequently detected in Mexico City. The Rt value for variant B.1.1.519 fluctuated strongly in the months following December, which may have been influenced by the small number of cases associated with this variant. This strong fluctuation differs from other variants detected in that region as other variants stabilized or disappeared as the year progressed.
The detailed maximum phylogeny was calculated, which included all SARS-CoV-2 genomes of interest to examine the geographic origin of variant B.1.1.519 and its evolutionary relationship to the B.1.1.222 variant.
There are three defined clusters shown in the phylogenetic tree. Two only correspond to the B.1.1.519 and B.1.1.222 variants, with clear separation and a mixed cluster, displaying undefined separation among lineages.
This demonstrates that the evolution of this SARS-CoV-2 variant lineage remains unclear, although the mixed cluster formed by variant B.1.1.519 sequences is most closely related to variant B.1.1.222 sequences.
The research team studied the clinical impact of the B.1.1.519 variant by analyzing associations between the variant and the number of clinical traits. The only sequences considered for the analyses were those with complete sets of clinical data (N=600).
Alarmingly evaluation of the data revealed that patients infected with the B.1.1.519 variant displayed a significant increase in the likelihood of developing symptoms affecting the respiratory tract relative to non-B.1.1.519 variants.
Detailed logistic regression models adjusted for viral cycle threshold, number of comorbidities, age, and sex revealed that B.1.1.519 variant was associated with a 1.786-fold increase in shortness of breath, a 1.489-fold increase in chest pain, and a 3.655-fold increase in cyanosis. Also, variant B.1.1.519 was associated with a higher fraction of patients developing serious illness or death.
The study team observed that the B.1.1.519 SAR-CoV-2 variant was significantly associated with hospitalization, severe disease, and death. Patients infected with variant B.1.1.519 also appeared to show a higher prevalence of symptoms such as shortness of breath, chest pains, and cyanosis.
It should be noted that the more severe outcomes associated with variant B.1.1.519 are similar to those shown with the delta variant. Recent research has shown that infections with the delta variant put patients at a higher risk of severe disease leading to hospitalization.
Despite reports sent to the WHO, the rather slow and complacent international health agency still has not elevated the B.1.1.519 variant to that of a Variant of Concern or VOC!
Mexican researchers are warning that the variant is still spreading globally and worse, new sub-variants of the B.1.1.591 with unique mutations and deletions are being found and some are worrying the research teams.
Both study teams warn that sustained genomic surveillance is critical to identify newly emerging variants. Any significant clinical associations could be of great importance to attempt to contain the pandemic.
In Latin America, with the exception of P.1 and P.2 observed in Brazil, no other variants with the potential for rapid expansion have been reported so far [11]. Here, we report the identification of a potential VOI harboring the mutations T478K, P681H, and T732A in the spike protein, within the newly named lineage B.1.1.519, derived from the B.1.1.222 lineage, that rapidly outcompeted the preexisting variants in Mexico and has been the dominant virus in the country during 2021.
Derived from genomic surveillance carried out in Mexico, 2,692 genomic sequences were obtained in this study and are part of the 3,156 sequences deposited in the GISAID from March 1, 2020 to March 21, 2021. As a result of the analysis of this set of sequences, we observed the presence of 91 Phylogenetic Assignment of Named Global Outbreak (PANGO) lineages, with B.1.1.519 (37.8%), B.1 (13.9%), B.1.1.222 (10.3%), B.1.1 (5.7%), B.1.609 (5.6%), and B.1.243 (4.5 %) being the most prevalent. Libraries for whole genome sequencing of SARS-CoV-2 were generated using the protocol developed by the ARTIC Network (https://artic.network/2-protocols.html) or a long-amplicon-based method (https://pubmed.ncbi.nlm.nih.gov/32222995/).
A striking observation was the detection of the B.1.1.519 lineage in the USA, derived from B.1.1.222, which harbors the mutation T478K in the spike protein. This variant had not been detected in Mexico before October 2020, when it was found in Mexico City, and phylogeographic analysis suggested that the B.1.1.519 variant emerged around mid-September 2020 [10].
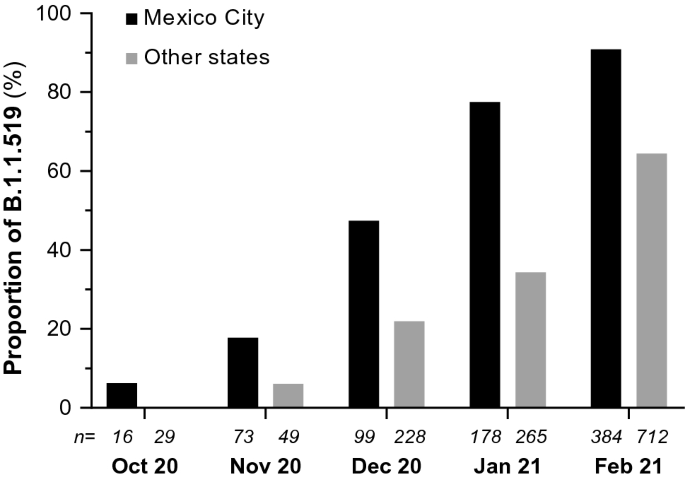
In November 2020, 13% (16/123) of the characterized cases of COVID-19 were caused by this variant, and in December, this proportion increased to 29.3% (97/331). In January 2021, the percentage of B.1.1.519 rose to 51.5% (229/445), increasing in incidence to 73.6% (808/1098) in February. On the other hand, a decreasing frequency of the B.1 lineage that had predominated in Mexico in 2020 was observed, going from 36.27% (284/783) between March and September to 2.37% (26/1098) in February 2021.
In Mexico, since the identification of B.1.1.519 in November 2020, a total of 6419 genomic sequences have been reported with this lineage. The majority of them are from Mexico City and are spread throughout all country.
A detailed analysis of the samples from Mexico City indicated that, in November, this variant was present in 17.8% (13/73) of the cases, while in December 2020, this proportion increased to 47.5% (47/99). In January 2021, the variant was detected in 77.5% (138/178) of the cases and by February in 90.9% (349/384). This significant increase in the frequency of B.1.1.519 in Mexico City showed that it outcompeted preexisting variants between October 2020 and February 2021, and this increase was also observed in other regions of the country (Fig. 1), representing more than 50% of the characterized viruses in some states during the first trimester of 2021. In particular, the variant was highly prevalent in Baja California Sur (51.3%, 20/39), Guerrero (70%, 21/30), Hidalgo (72.2%, 13/18), Morelos (67.3%, 33/49), State of Mexico (83.5%, 76/91), Oaxaca (51.3%, 19/37), Puebla (78.5%, 77/98), Queretaro (70.8%, 34/48), San Luis Potosi (70%, 35/50), and Veracruz (69.5%, 80/115).
This variant has also been detected in 17 countries on all five continents. In the Americas, it has been reported in Canada and the USA, and recently in Brazil, Chile, Aruba, Martinique, and Curazao [12]. However, this variant currently is not predominant in these countries.
The overall genome analysis of the viruses in the B.1.1.519 lineage showed the presence of 20 mutations in total, compared to the Wuhan-Hu-1 reference genome sequence (NCBI accession number MN908947). Eleven of these mutations are non-synonymous, and four of them are present in the spike protein. Notably, a T478K mutation is present in the receptor binding domain (RBD), where mutations have been shown to reduce the activity of some monoclonal antibodies [9]. All amino acid and nucleotide changes are listed in Table 1.
Table 1 Amino acid and nucleotide changes
| Mutations | Gene | |
|---|---|---|
| Nucleotide | Amino acid | |
| C203T | – | |
| C222T | – | |
| C241T | – | |
| C3037T | – | ORF1a |
| C3140T | P141S | |
| C10029T | T492I | |
| C10954T | – | |
| A11117G | I49V | |
| C12789T | ||
| C14408T | P323L | ORF1b |
| T19839C | – | |
| C21306T | – | |
| C22995A | T478K | Spike |
| A23403G | D614G | |
| C23604A | P681H | |
| A23756G | T732A | |
| G28881A | – | N |
| G28882A | R203K | |
| G28883C | G204R | |
| C29197T | – |
- The main amino acid substitutions are shown in bold
The current B.1.1.519 lineage, represented by the vast majority of the reported Mexican sequences, was first identified as a B.1.1.222 lineage. However, the presence of the mutations T478K, P681H, and T732A clearly differentiated it from this lineage, which does not contain these mutations, giving rise to the B.1.1.519 lineage. A phylogenomic analysis of genomic sequences using the Nextstrain tool showed that the viruses in the lineage B.1.1.519 (B.1.1.1.222+T478K+P681H+T732A) group independently of the lineage B.1.1.222 sequences, strongly suggesting that this variant should be classified as a variant of interest (VOI) (Fig. 2). On the other hand, viruses in the lineage B.1.1.519 are already grouped by the GISAID platform in an independent clade, invariably harboring the three mutations mentioned above.
An in silico analysis using different potent structures of related strains suggested that the position of the T478K mutation in the S protein is involved in antibody recognition and the receptor binding site [13]. In a deep mutational scanning of the SARS-CoV-2 receptor binding domain, the T478K mutation did not have a significant effect on folding or binding to human angiotensin-converting enzyme 2 (ACE2) [14].
However, this mutation may be involved in immune evasion, particularly escape from antibody neutralization [15]. The P681H mutation is one of the mutations found in the B.1.1.7 variant detected in the UK. According to the definitions described in the document issued by the WHO “Covid-19 Weekly Epidemiological Update” of February 25, 2021, with the special edition of “Proposed Working Definitions of SARS-CoV-2 Variants of Interest and Variants of Concern”, we can consider the lineage B.1.1.519 a potential variant of interest [16].
Finally, two variants with interesting features were identified in this study: first, 13 sequences belonging to the B.1.1.222 lineage without the T478K mutation, but harboring the T732A mutation and the 69-70 deletion in the spike protein, the latter being a characteristic mutation of the B.1.1.7 VOC first detected in the UK; and second, 11 sequences corresponding to four lineages differing from B.1.1.519 (B.1, B.1.1.222, B.1.1.322, and B.1.323) but containing the same T478K, P681H, and T732A mutations in the spike glycoprotein that are present in the variant B.1.1.519. Keeping track of the incidence of these two variants is recommended during genomic surveillance.
So far, we do not have experimental evidence to determine if the mutations described here could be associated with changes in transmission, virulence, and/or antigenicity or if they could have an impact on the severity of disease, reinfection rates, or vaccine effectiveness. For this reason, the importance of a genomic surveillance system, epidemiological studies, and experiments to assess the neutralization of viruses in lineage B.1.1.519 or any new variants are crucial for investigating the possible biological impact of the mutations in the context of public health. Fortunately, all COVID-19 virus variants that have emerged so far respond to the available, approved vaccines to some extent.
reference link :https://link.springer.com/article/10.1007/s00705-021-05208-6



{kind=link}