









 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Un nuovo studio condotto da ricercatori tedeschi del Center for Infection and Genomics of the Lung (CIGL), Università di Giessen e Marburg Lung Center, Giessen-Germania, insieme a scienziati di numerosi altri istituti di ricerca tedeschi, ha sorprendentemente scoperto che il SARS-CoV- 2 induce la formazione di strutture di coagulo di fibrina uniche e diverse e mai viste prima nel corpo umano e sono queste strutture alterate del coagulo di fibrina che stanno contribuendo al rischio di trombosi nei pazienti gravi di COVID-19.
Secondo l’abstract dello studio, “L’elevata incidenza di eventi trombotici suggerisce un possibile ruolo del percorso del sistema di contatto nella patologia COVID-19”.
Il team di studio ha dimostrato livelli alterati di fattore XII (FXII) e dei suoi prodotti di attivazione in due coorti indipendenti di pazienti critici COVID-19 rispetto ai pazienti affetti da sindrome da distress respiratorio acuto grave dovuta al virus dell’influenza (ARDS-influenza).
Il team di studio ha riscontrato un rapido consumo di FXII nel plasma COVID-19, ma non nell’influenza ARDS.
È interessante notare che il tempo di coagulazione del caolino non è stato prolungato nel COVID-19 rispetto all’influenza ARDS.
Utilizzando la microscopia confocale ed elettronica, il team di studio ha dimostrato che l’aumento del tasso di attivazione del FXII, insieme a livelli elevati di fibrinogeno, la formazione di fibre resistenti alla fibrinolisi, coaguli compatti con fibre sottili e piccoli pori nel COVID-19.
Il team di studio ha anche osservato la lisi del coagulo nel 30% dei pazienti COVID-19 e nell’84% dei soggetti con influenza ARDS. L’analisi delle sezioni di tessuto polmonare ha rivelato depositi di fibrina compatta extra e intra-vascolare diffusa in COVID-19.
I risultati dello studio indicano che livelli elevati di fibrinogeno e un aumento del tasso di attivazione del FXII promuovono la trombosi e la resistenza alla trombolisi attraverso una maggiore formazione e stabilità di trombi nel COVID-19.
I risultati dello studio sono stati pubblicati su un server di prestampa e sono attualmente in fase di revisione paritaria. https://www.biorxiv.org/content/10.1101/2021.09.17.460777v1
Il coronavirus SARS-CoV-2 causa la malattia COVID-19, che è una malattia multisistemica che colpisce le vie respiratorie. I dati finora disponibili mostrano il danno endoteliale causato dall’iperattivazione del sistema immunitario come uno dei principali meccanismi molecolari alla base della gravità e della mortalità del COVID-19.
Inoltre, questo coinvolgimento endoteliale porta a molte anomalie dell’emostasi nei pazienti gravi con COVID-19.
Oltre a livelli elevati di mediatori pro e antinfiammatori come interleuchina (IL)-6, IL-10, IL-2R e fattore di necrosi tumorale-α (TNF-α), aumentati livelli di D-dimero, fibrinogeno e tempo di protrombina (PT) prolungato è stato riscontrato anche in pazienti affetti da COVID-19 gravi.
L’osservazione e il monitoraggio di questi biomarcatori elevati sono clinicamente rilevanti perché livelli anormali di D-dimero sono associati a mortalità a 28 giorni nei pazienti COVID-19.
Inoltre, studi post mortem mostrano la presenza di microtrombi e capillarostasi nei polmoni di questi gravi pazienti affetti da COVID-1. L’elevata incidenza di eventi trombotici nei pazienti affetti da COVID-19 grave indica un possibile ruolo del percorso del sistema di contatto nella patologia COVID-19.
Il team di ricerca COVID-19 ha dimostrato che livelli alterati di fattore XII (FXII) e dei suoi prodotti di attivazione in 2 coorti indipendenti di pazienti critici con COVID-19 rispetto ai pazienti affetti da sindrome da distress respiratorio acuto grave causata dal virus dell’influenza (acuta respiratoria sindrome da distress (ARDS)-influenza).
Il fattore XII della coagulazione, noto anche come fattore di Hageman o semplicemente FXII, è una proteina plasmatica. È la forma zimogena del fattore XIIa, un enzima (EC 3.4.21.38) della classe della serina proteasi (o serina endopeptidasi). Nell’uomo, il fattore XII è codificato dal gene F12. Tipicamente circola nel plasma come zimogeno a catena singola.
Entrando in contatto con superfici anioniche come caolino, trappole extracellulari dei neutrofili, RNA extracellulare da cellule danneggiate o polifosfati da piastrine attivate, FXII viene autoattivato in αFXIIa.
È importante sottolineare che l’attivazione del sistema della fase di contatto porta ad un aumento della produzione di trombina e fibrina, sebbene la conversione del plasminogeno in plasmina mediata da FXIIa/PKa possa avere un lieve effetto sulla fibrinolisi.
Il team di studio ha anche riportato un rapido consumo di FXII nel plasma dei pazienti COVID-19, ma non nel plasma dell’influenza ARDS, che è compatibile con i dati di cui sopra. È interessante notare che il tempo di coagulazione del caolino non è stato prolungato nei pazienti COVID-19 rispetto a quello nei pazienti con influenza ARDS.
L’autore corrispondente, la dott.ssa Malgorzata Wygrecka, PhD del Center for Infection and Genomics of the Lung (CIGL) University of Giessen e del Marburg Lung Center, ha dichiarato al Thailand Medical News: “È stato riportato che livelli elevati di fibrinogeno contribuiscono alla formazione più rapida di fibrina e all’aumento della rete di fibrina. densità, forza e stabilità”.
Utilizzando la microscopia confocale ed elettronica, il team di studio ha dimostrato che l’aumento del tasso di attivazione del FXII, insieme a livelli elevati di fibrinogeno, guida la formazione di coaguli resistenti alla fibrinolisi mai visti prima con fibre sottili e piccoli pori nei pazienti COVID-19.
Il dott. Wygrecka ha commentato: “I coaguli generati dal plasma COVID-19 hanno mostrato una maggiore densità di impacchettamento, pori piccoli ed erano costituiti da fibre sottili”.
I risultati dello studio hanno rivelato che la lisi del coagulo è stata osservata nel 30% dei pazienti con COVID-19 e nell’84% dei pazienti con influenza ARDS.
È importante sottolineare che dopo un’analisi dettagliata delle sezioni di tessuto polmonare dei pazienti COVID-19, sono stati rivelati depositi di fibrina compatta extra e intra-vascolare estesi ma unici.
I risultati dello studio aiutano a stabilire un modello per studi futuri sul ruolo della struttura alterata del coagulo di fibrina nella trombosi correlata al COVID-19.
Secondo il team di studio, sulla base dei risultati, la risposta incontrollata dei meccanismi di difesa, compreso il sistema immunitario e di coagulazione, costituisce il meccanismo alla base dell’infezione grave da SARS-CoV-2.
È importante sottolineare che le anomalie nella composizione del plasma, le cellule immunitarie del sangue dovute al danno cellulare mediato da virus e il rilascio di detriti intracellulari favoriscono l’attivazione di FXII.
Oltre agli alti livelli di fibrinogeno, FXIIa porta alla formazione patologica di trombi non solo attraverso la generazione di trombina ma anche attraverso la formazione di coaguli compatti e resistenti alla lisi.
I risultati dello studio indicano che l’aumento dei livelli di fibrinogeno e l’aumento del tasso di attivazione del FXII guidano la trombosi e la resistenza alla trombolisi attraverso una maggiore formazione e stabilità di trombi nei pazienti COVID-19.
Il team di studio ha concluso: “Questo studio stabilisce quindi un modello per studi futuri sul ruolo della struttura alterata del coagulo di fibrina nella trombosi e nella trombolisi in pazienti gravi con COVID-19. Se l’interazione di FXII/FXIIa con il fibrinogeno può interferire con il legame del t-PA con la fibrina e quindi inibire la fibrinolisi, merita ulteriori indagini».
L’infezione indotta da sindrome respiratoria acuta grave da coronavirus 2 (SARS-Cov-2), causa della malattia da coronavirus 2019 (COVID-19), è caratterizzata da patologie cliniche senza precedenti. Le caratteristiche fenotipiche vascolari sono fortemente associate a varie coagulopatie che possono provocare sanguinamento e trombocitopenia o ipercoagulazione e trombosi (Gupta et al., 2020, Perico et al., 2021).
Vari biomarcatori della coagulazione infiammatoria circolanti e disregolati, tra cui fibrina (ogeno), D-dimero, P-selectina e fattore di von Willebrand (VWF), proteina C-reattiva (CRP) e varie citochine, si legano direttamente ai recettori endoteliali. Le endoteliopatie sono quindi una caratteristica clinica chiave della condizione (Goshua et al., 2020, Ackermann et al., 2020).
Durante la progressione delle varie fasi del COVID-19, è probabile che i marcatori della replicazione virale, nonché la deplezione di VWF e fibrinogeno con livelli aumentati di D-dimero e livelli di P-selectina disregolati, seguiti da una tempesta di citochine, siano indicativi di una prognosi sfavorevole (Grobler et al., 2020, Pretorius et al., 2020, Venter et al., 2020, Roberts et al., 2020).
Questa prognosi infausta è ulteriormente peggiorata poiché insieme a una sostanziale deposizione di microcoaguli nei polmoni (Renzi et al., 2020, Ciceri et al., 2020, Bobrova et al., 2020), il plasma dei pazienti COVID-19 porta anche un massiccio carico di coaguli amiloidi preformati (Grobler et al., 2020), e ci sono anche numerose segnalazioni di danni agli eritrociti (Lam et al., 2020, Berzuini et al., 2020, Akhter et al., 2020), piastrine e disregolazione di biomarcatori infiammatori (Grobler et al., 2020, Pretorius et al., 2020, Venter et al., 2020, Roberts et al., 2020).
La virulenza del patogeno è strettamente legata alle sue proteine di membrana. Una di queste proteine, che si trova sul virus COVID-19, è la proteina spike, che è una glicoproteina di membrana. Le proteine spike sono i fattori chiave per l’attaccamento del virus alle cellule bersaglio, poiché si legano ai recettori di superficie che convertono l’angiotensina 2 (ACE2) (Bergmann e Silverman, 2020). Le proteine Spike sono proteine di fusione virale di classe I (Kawase et al., 2019).
Si presentano come omotrimeri sporgenti sulla superficie virale e mediano l’ingresso del virus nelle cellule ospiti bersaglio (Walls et al., 2020). Una singola proteina spike ha una dimensione compresa tra 180 e 200 kDa e contiene un N-terminale extracellulare, un dominio transmembrana fissato nella membrana del virus e un breve segmento C-terminale intracellulare (Kawase et al., 2019, Zhang et al. ., 2020). Le proteine spike sono rivestite con molecole di polisaccaridi che fungono da camuffamento.
Questo aiuta a eludere la sorveglianza da parte del sistema immunitario dell’ospite durante l’ingresso (Zhang et al., 2020). La subunità S1 è responsabile del legame al recettore (Watanabe et al., 2020), con la subunità 2 (S2), una subunità carbossi-terminale, responsabile della fusione e dell’ingresso virale (Flores-Alanis et al., 2020) (vedi Figura 1 ).
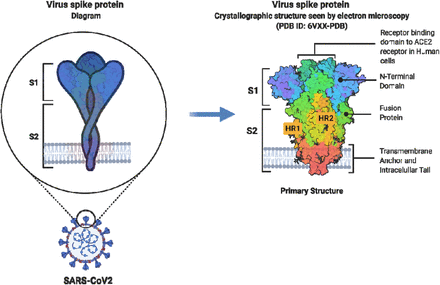
Rappresentazione schematica della glicoproteina Spike SARS-CoV-2. Adattato da (Duan et al., 2020). Abbreviazioni: S1, subunità 1; S2, subunità 2; HR1, ripetizione eptade 1; HR2, ripetizione eptade 2. Questa immagine è stata creata con BioRender (https://biorender.com/).
Il legame al recettore è certamente responsabile delle patologie cellulo-mediate, ma non spiega di per sé le coagulopatie. La proteina Spike, tuttavia, può essere eliminata ed è stata rilevata in vari organi, compreso il tratto urinario (George et al., 2021). Le proteine S1 possono anche attraversare la barriera ematoencefalica (Rhea et al., 2021). Le particelle S1 libere possono anche svolgere un ruolo nella patogenesi della malattia (Letarov et al., 2020, Buzhdygan et al., 2020). La proteina spike libera può potenzialmente essere rilasciata a causa dello “scatto” spontaneo dei trimeri della proteina S sulla superficie dei virioni e le cellule infette liberano particelle S1 contenenti il dominio legante il recettore libero (Letarov et al., 2020).
Qui studiamo l’effetto della subunità S1 della proteina spike isolata SARS-CoV-2 come potenziale infiammatorio pro-infiammatorio sui generis. Indaghiamo il potenziale di questo inflammagen di interagire direttamente con le piastrine e la fibrina (ogeno) per causare cambiamenti della proteina fibrina (ogeno) e ipercoagulazione del sangue. Determiniamo anche se la proteina spike può interferire con il flusso sanguigno, confrontando campioni PPP sani naïve, con e senza proteina spike aggiunta, con campioni PPP di pazienti positivi al COVID-19 (prima del trattamento). Concludiamo che la proteina spike può avere effetti patologici direttamente, senza essere assorbita dalle cellule. Ciò fornisce ulteriori prove del fatto che il bersagliarlo direttamente, tramite vaccini o anticorpi, potrebbe essere di beneficio terapeutico.
collegamento di riferimento: https://www.medrxiv.org/content/10.1101/2021.03.05.21252960v1.full



{kind=link}