










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



A new study led by German researchers from the Center for Infection and Genomics of the Lung (CIGL), Universities of Giessen and Marburg Lung Center, Giessen-Germany along with scientist from numerous other German research institutions have shockingly found that the SARS-CoV-2 induces the formation of unique and different and fibrin clot structures never seen before in the human body and it is these altered fibrin clot structures that are contributing to thrombosis risk in severe COVID-19 patients.
According to the study abstract, “The high incidence of thrombotic events suggests a possible role of the contact system pathway in COVID-19 pathology.”
The study team demonstrated altered levels of factor XII (FXII) and its activation products in two independent cohorts of critically ill COVID-19 patients in comparison to patients suffering from severe acute respiratory distress syndrome due to influenza virus (ARDS-influenza).
The study team found rapid consumption of FXII in COVID-19, but not in ARDS-influenza, plasma.
Interestingly, the kaolin clotting time was not prolonged in COVID-19 as compared to ARDS-influenza.
Utilizing confocal and electron microscopy, the study team showed that increased FXII activation rate, in conjunction with elevated fibrinogen levels, triggers formation of fibrinolysis-resistant, compact clots with thin fibers and small pores in COVID-19.
The study team also observed clot lysis in 30% of COVID-19 patients and 84% of ARDS-influenza subjects. Analysis of lung tissue sections revealed wide-spread extra- and intra-vascular compact fibrin deposits in COVID-19.
The study findings indicate that elevated fibrinogen levels and increased FXII activation rate promote thrombosis and thrombolysis resistance via enhanced thrombus formation and stability in COVID-19.
The study findings were published on a preprint server and are currently being peer reviewed. https://www.biorxiv.org/content/10.1101/2021.09.17.460777v1
The SARS-CoV-2 coronavirus causes the COVID-19 disease, which is a multisystem disease affecting the respiratory tract. Data available so far shows endothelial injury caused by hyperactivation of the immune system as a major underlying molecular mechanism for COVID-19 severity and mortality.
In addition, this endothelial involvement leads to many hemostasis abnormalities in severe COVID-19 patients.
Besides elevated levels of pro- and anti-inflammatory mediators such as interleukin (IL)-6, IL-10, IL-2R, and tumor necrosis factor-α (TNF-α), increased levels of D-dimer, fibrinogen, and prolonged prothrombin time (PT) have also been found in severe COVID-19 patients.
The observation and monitoring of these elevated biomarkers are clinically relevant because abnormal levels of D-dimer are associated with 28-day mortality in COVID-19 patients.
Furthermore post-mortem studies show the presence of micro-thrombi and capillarostasis in the lungs of these severe COVID-1 patients. The high incidence of thrombotic events in severe COVID-19 patients in dicates a possible role of the contact system pathway in COVID-19 pathology.
The COVID-19 Research team demonstrated that altered levels of factor XII (FXII) and its activation products in 2 independent cohorts of critically ill patients with COVID-19 compared to patients suffering from severe acute respiratory distress syndrome caused by the influenza virus (acute respiratory distress syndrome (ARDS)-influenza).
Coagulation factor XII, also known as Hageman factor or simply FXII, is a plasma protein. It is the zymogen form of factor XIIa, an enzyme (EC 3.4.21.38) of the serine protease (or serine endopeptidase) class. In humans, factor XII is encoded by the F12 gene. It typically circulates in plasma as a single-chain zymogen.
Upon coming into contact with anionic surfaces like kaolin, neutrophil extracellular traps, extracellular RNA from damaged cells, or polyphosphates from activated platelets, FXII is autoactivated to αFXIIa.
Importantly activation of the contact-phase system leads to increased thrombin and fibrin production, although FXIIa/PKa-mediated conversion of plasminogen to plasmin may have a mild effect on fibrinolysis.
The study team also reported rapid consumption of FXII in the plasma of COVID-19 patients, but not in plasma from ARDS-influenza, which is compatible with the above data. Interestingly, the kaolin clotting time was not prolonged in COVID-19 patients compared to that in ARDS-influenza patients.
Corresponding author Dr Malgorzata Wygrecka, PhD from the Center for Infection and Genomics of the Lung (CIGL) Universities of Giessen and Marburg Lung Center told Thailand Medical News, “Elevated levels of fibrinogen were reported to contribute to the faster fibrin formation and increased fibrin network density, strength, and stability.”
Utilizing confocal and electron microscopy, the study team showed that increased FXII activation rate, along with elevated fibrinogen levels, drives the formation of unique never seen before fibrinolysis-resistant clots with thin fibers and small pores in COVID-19 patients.
Dr Wygrecka commented, “Clots generated from COVID-19 plasma exhibited higher packing density, small pores and were built of thin fibers.”
The study findings revealed that clot lysis was observed in 30% of COVID-19 patients and 84% of ARDS influenza patients.
Importantly upon detailed analysis of lung tissue sections of COVID-19 patients, extensive but unique extra- and intra-vascular compact fibrin deposits were revealed.
The study findings help establish a model for future studies on the role of altered fibrin clot structure in COVID-19-related thrombosis.
According to the study team, on the basis of the findings, the uncontrolled response of defense mechanisms, including the immune and coagulation system, constitute the underlying mechanism for severe SARS-CoV-2 infection.
Importantly abnormalities in plasma composition, blood immune cells due to virus-mediated cell damage, and release of intracellular debris all favor the activation of FXII. Apart from high levels of fibrinogen, FXIIa leads to pathologic thrombus formation not only through thrombin generation but also via the formation of compact and lysis-resistant clots.
The study findings indicate that increased fibrinogen levels and increased FXII activation rate drive thrombosis and thrombolysis resistance through enhanced thrombus formation and stability in COVID-19 patients.
The study team concluded, “Thus, this study establishes a model for future studies on the role of altered fibrin clot structure in thrombosis and thrombolysis in severe COVID-19 patients. Whether the interaction of FXII/FXIIa with fibrinogen can interfere with the binding of t-PA to fibrin and thereby inhibits fibrinolysis warrants further investigation.”
Severe acute respiratory syndrome coronavirus 2 (SARS-Cov-2)-induced infection, the cause of coronavirus disease 2019 (COVID-19), is characterized by unprecedented clinical pathologies. Phenotypic vascular characteristics are strongly associated with various coagulopathies that may result in either bleeding and thrombocytopenia or hypercoagulation and thrombosis (Gupta et al., 2020, Perico et al., 2021).
Various circulating and dysregulated inflammatory coagulation biomarkers, including fibrin(ogen), D-dimer, P-selectin and von Willebrand Factor (VWF), C-reactive protein (CRP), and various cytokines, directly bind to endothelial receptors. Endotheliopathies are therefore a key clinical feature of the condition (Goshua et al., 2020, Ackermann et al., 2020).
During the progression of the various stages of the COVID-19, markers of viral replication, as well as VWF and fibrinogen depletion with increased D-dimer levels and dysregulated P-selectin levels, followed by a cytokine storm, are likely to be indicative of a poor prognosis (Grobler et al., 2020, Pretorius et al., 2020, Venter et al., 2020, Roberts et al., 2020).
This poor prognosis is further worsened as together with a substantial deposition of microclots in the lungs (Renzi et al., 2020, Ciceri et al., 2020, Bobrova et al., 2020), plasma of COVID-19 patients also carries a massive load of preformed amyloid clots (Grobler et al., 2020), and there are also numerous reports of damage to erythrocytes (Lam et al., 2020, Berzuini et al., 2020, Akhter et al., 2020), platelets and dysregulation of inflammatory biomarkers (Grobler et al., 2020, Pretorius et al., 2020, Venter et al., 2020, Roberts et al., 2020).
The virulence of the pathogen is closely linked to its membrane proteins. One such protein, found on the COVID-19 virus, is the spike protein, which is a membrane glycoprotein. The spike proteins are the key factors for virus attachment to target cells, as they bind to the angiotensin-converting 2 (ACE2) surface receptors (Bergmann and Silverman, 2020). Spike proteins are class I viral fusion proteins (Kawase et al., 2019).
They present as protruding homotrimers on the viral surface and mediate virus entry into the target host cells (Walls et al., 2020). A singular spike protein is between 180–200 kDa in size and contains an extracellular N-terminal, a transmembrane domain fixed in the membrane of the virus, and a short intracellular C-terminal segment (Kawase et al., 2019, Zhang et al., 2020). Spike proteins are coated with polysaccharide molecules that serve as camouflage.
This helps evade surveillance by the host immune system during entry (Zhang et al., 2020). The S1 subunit is responsible for receptor binding (Watanabe et al., 2020), with subunit 2 (S2), a carboxyl-terminal subunit, responsible for viral fusion and entry (Flores-Alanis et al., 2020) (see Figure 1).
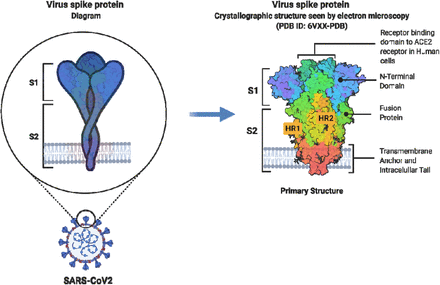
Schematic representation of SARS-CoV-2 Spike glycoprotein. Adapted from (Duan et al., 2020). Abbreviations: S1, subunit 1; S2, subunit 2; HR1, heptad repeat 1; HR2, heptad repeat 2. This image was created with BioRender (https://biorender.com/).
Receptor binding is certainly responsible for cell-mediated pathologies, but does not of itself explain the coagulopathies. Spike protein, can however be shed, and it has been detected in various organs, including the urinary tract (George et al., 2021). S1 proteins can also cross the blood-brain-barrier (Rhea et al., 2021). Free S1 particles may also play a role in the pathogenesis of the disease (Letarov et al., 2020, Buzhdygan et al., 2020). Free spike protein can potentially be released due to spontaneous “firing” of the S protein trimers on the surface of virions, and infected cells liberates free receptor binding domain-containing S1 particles (Letarov et al., 2020).
Here we study the effect of isolated SARS-CoV-2 spike protein S1 subunit as potential pro-inflammatory inflammagen sui generis. We investigate the potential of this inflammagen to directly interact with platelets and fibrin(ogen) to cause fibrin(ogen) protein changes and blood hypercoagulation. We also determine if the spike protein may interfere with blood flow, by comparing naïve healthy PPP samples, with and without added spike protein, to PPP samples from COVID-19 positive patients (before treatment). We conclude that the spike protein may have pathological effects directly, without being taken up by cells. This provides further evidence that targeting it directly, whether via vaccines or antibodies, is likely to be of therapeutic benefit.
reference link : https://www.medrxiv.org/content/10.1101/2021.03.05.21252960v1.full



{kind=link}
[…] SARS-CoV-2 Alters Fibrin Clot Structures That Contributes To Thrombosis Risk In… […]