











 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



This is revealed in a new study by researchers at Bielefeld University’s Center for Biotechnology (CeBiTec) and the Evangelical Hospital Bethel (EvKB), one of the supporting hospitals of Medical School OWL.
The research team was able to show that a large part of the ORF8 gene segment was missing in the samples it analyzed. This gene region is thought to contribute to delaying defensive reactions in the human body. If it is missing, there is a chance that the virus will become less pathogenic, meaning that it will cause less serious disease. The research team has published its findings in the journal Viruses.
“When both hospitals and schools carry out routine tests to see whether people have caught the virus, their main aim is to contain further infections,” says lead investigator Professor Dr. Jörn Kalinowski, a geneticist at CeBiTec.
When laboratories analyze samples with the widely available PCR tests, they are not just aiming to determine whether an infection is present. If they find a case of infection, they also investigate which variant of the virus is involved.
“To do this, it is sufficient to identify individual characteristic gene sections that are typical for the common virus variants.” Currently, such analyses usually produce the same result throughout Europe: the delta variant – it is far more infectious than other variants.
“Because only a few gene segments are required to identify a common viral variant, laboratories usually simply accept that they cannot identify other gene segments,” says Kalinowski. One reason why the genome cannot be determined completely is, for example, inadequate preparation of the sample.
In addition, however, it is often the case that analysis software does not optimally recognize individual nucleotides – the gene building blocks of the viral genome. Instead of the letters A, T, G, and C, which are used to represent the gene building blocks of the viral genome, the software then writes the letter N into the gene sequence.
Existing software documents missing gene segments in a misleading way
The Bielefeld research team’s study found that this makeshift approach can lead to a far-reaching problem. “Mutations can lead to variants of SARS-CoV-2 in which longer gene segments are deleted,” says bioinformatician Professor Dr. Alexander Sczyrba from CeBiTec, co-author of the study.
“We found that the commonly used standard software enters placeholders in the gene sequence even when an entire gene segment is not present at all.” Then the letter N is written in rows in the gene sequence.
“This is a systematic error,” says Jörn Kalinowski, ‘because such a deletion in the genome is an important clue when it comes to future exposure to the coronavirus.” When there is a deletion in a gene segment, properties stored in the affected gene also disappear. As the virus replicates, these properties are no longer passed on.
“In addition, mutations that make the virus more dangerous for humans can no longer develop within such deletions in the genome.” According to Kalinowski, such missing gene segments can be one of the reasons why SARS-CoV-2 adapts to humans as its hosts. This would then make the virus more infectious, but, at the same time, less dangerous.
“The virus would then become endemic. In other words, it would appear regularly in different regions, as is the case with other, long-known coronaviruses that nowadays give us only harmless colds.”
Knowing the virus variant helps in reconstructing infection chains
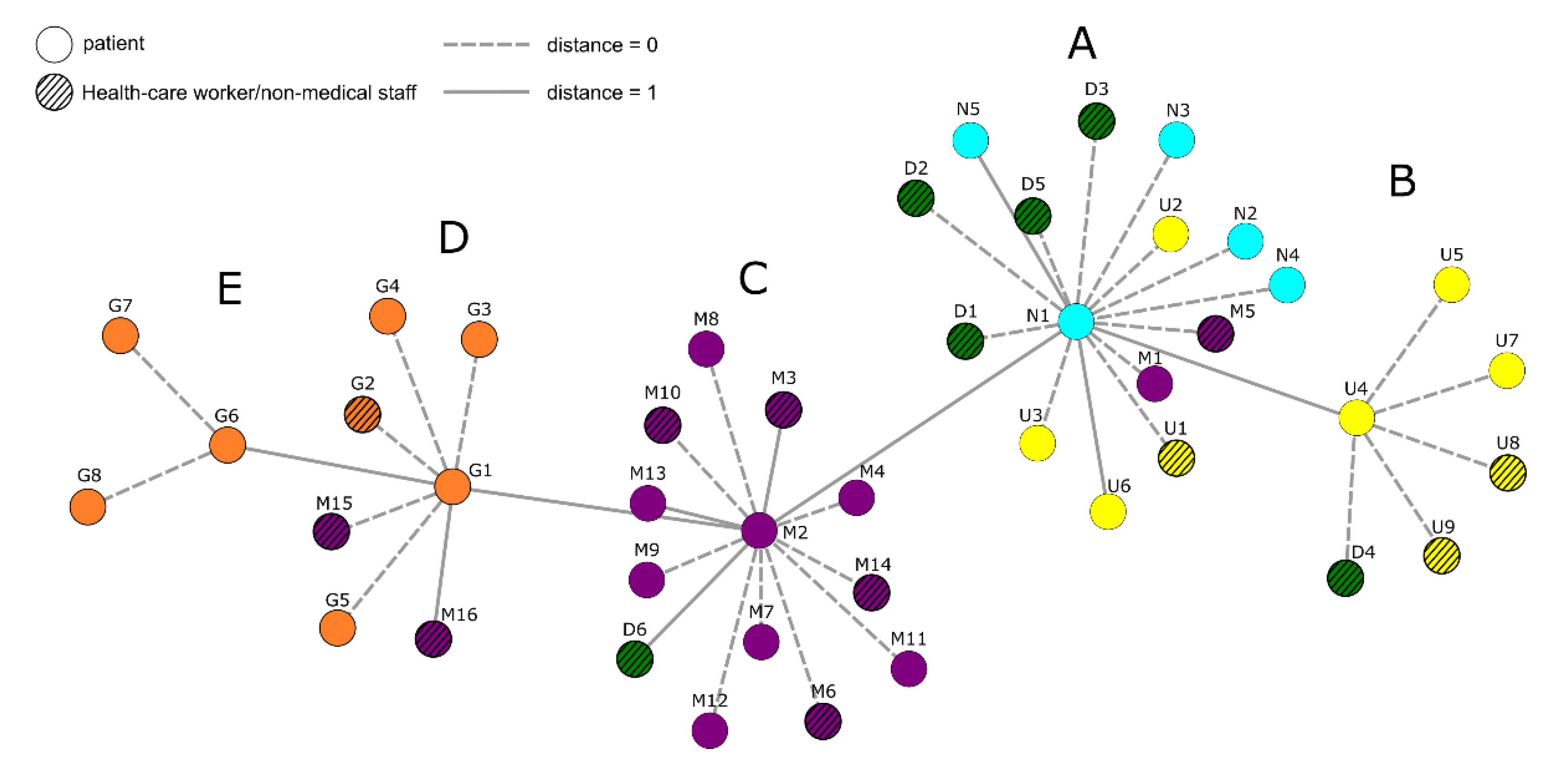
The researchers discovered the gene deletion in the virus when analyzing virus samples from the EvKB. There, medical staff and patients are tested continuously for infections with the coronavirus. Kalinowski’s research group has been analyzing samples from the hospital since April 2020.
For their analyses, they receive extracts containing the genetic material of the virus. These come from swabs of positively tested patients. “Detailed analysis of the samples enables us to reconstruct infection chains when cases occur,” says senior physician Dr. Christiane Scherer, head of microbiology at the EvKB and also co-author of the study.
At the peak of the second wave of infections in January and February 2021, Scherer and her team identified a cluster of infections. They managed to contain these through extensive screening and contact tracing. The viral variant B.1.1.294 was unable to spread further on the wards.
The CeBiTec analyses confirmed that the isolation measures on the affected wards were successful. “We were able to state this so precisely because we discovered a special feature of the virus variant: there are 168 nucleotides missing in its genetic code,” reports Jörn Kalinowski. The gene building blocks were missing in the gene region “Open Reading Frame 8′ (ORF8). This genetic information is presumably responsible for the fact that the virus succeeds in delaying the immune response of infected individuals.
Function added to analysis software
The scientists were able to detect the missing nucleotides because – in contrast to the standard PCR testing – they also applied nanopore sequencing. Compared to the usual sequencing machines, these special instruments make it possible to determine longer gene segments.
The researchers also added a further function to freely available gene analysis software that correctly detects and labels missing nucleotides in gene sequences. “This was the only way we could determine that part of the ORF8 gene region had disappeared,” says Kalinowski.
“This analysis allowed us not only to determine the cluster in our hospital,” says Christiane Scherer, ‘but also to ensure that the viral variant had reached a dead end with us and that no one else would contract it after it had been contained.”
Evolutionary dendrogram shows related variants of the investigated virus mutant
The CeBiTec researchers wanted to know the origin of the virus variant with the deleted gene segment. To do this, they took raw data from the central database of coronavirus variants and evaluated it using their own specially developed software. “We were thus able to determine where other predecessors of the variant we were investigating had emerged and where similar variants could be found,” explains Alexander Sczyrba.
The CeBiTec researchers also want to enable other scientists to precisely identify missing gene segments in SARS-CoV-2 variants. To this end, they are making their further development of the analysis software, including the source code, available for downloading on a relevant platform.
“If we want to clarify what are the functions of individual genes in the virus and how it evolves, it is important for us to be able to search for further variants with deleted gene segments,” says Jörn Kalinowski.
“But this would require access to all the raw data from analyzed coronavirus samples nationwide. Unfortunately, rigid data protection regulations currently prevent this,” he says.
In December 2019, a previously unknown coronavirus causing severe cases of pneumonia started to spread from the Hubei province in China and was later termed severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) due to its relatedness to SARS-CoV [1,2]. SARS-CoV-2 rapidly reached all continents and has caused over 120 million infections and 2.6 million deaths worldwide until March 2021 [3].
Coping with increasing human immunity, improved medical treatment strategies and vaccination efforts, mutations increasing viral fitness are being positively selected. To track and monitor viral genetic diversity, viral whole-genome sequencing (WGS) is widely employed and also partly mandated by public health authorities [4].
Here, targeted tiled-amplicon sequencing, e.g., using the ARTIC-ONT protocol [5], has been proven to be a key technology for WGS especially for low viral loads samples [6].
SARS-CoV-2 ORF8 codes for an accessory protein, which is located in a hypervariable region of about 430 nt common to several SARS-similar coronaviruses and is possibly involved in immune system evasion by downregulating MHC-I as well as inhibiting Type I interferon signaling [7,8,9,10]. Several genomic deletions in ORF8, ranging from 1 to 382 nt, have been reported for SARS-CoV-2 [10,11].
It was assumed that hairpins in the ORF8 transcript region could play a role in genomic rearrangements during virus replication [11]. The so-called ∆382 variant, which was first identified in Singapore in early 2020, apparently causes infections with milder clinical symptoms [12,13]. In addition, during the SARS-CoV epidemic in 2003, a 29 nt deletion in ORF8 splitting it into ORF8a and ORF8b led to decreased viral fitness and may have reduced the severity of the epidemic [14].
Genomic surveillance of SARS-CoV-2 is of great importance, but besides identification and tracking of variants of concern (VOCs), which potentially pose the risks of higher infectivity or immune system evasion, the systematic screening for deletion mutants is also of high interest in order to monitor their spread and assess their impact on the severity of the COVID-19 disease as well as immunity in populations.
reference link :https://www.mdpi.com/1999-4915/13/9/1870/htm
More information: David Brandt et al, Multiple Occurrences of a 168-Nucleotide Deletion in SARS-CoV-2 ORF8, Unnoticed by Standard Amplicon Sequencing and Variant Calling Pipelines, Viruses (2021). DOI: 10.3390/v13091870



{kind=link}