










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



They published their paper in the March issue of Journal of Biological Chemistry.
Serious gum infections damage soft mouth tissues such as gums and gradually erode the underlying (alveolar) bones that support our teeth. Both the bone pockets around the base of teeth and the ligaments anchoring teeth to the jawbone are susceptible to getting broken away by bacterial infection. This periodontal bone erosion, gone unchecked, may finally result in tooth loss.
Lipopolysaccharides support the bacterial cell and protect against attack of immune cells, but have also been implicated in causing gum inflammation by switching on toll-like receptors (TLR4) on immune cells that then recognize the bacteria as pathogens.
For example, immune cells such as neutrophils accumulated in inflammatory tissues could release dsRNA in the mouth. The recent study investigated dsRNA as a suspect in the progression of bone inflammation during periodontal disease.
In healthy bones, stromal osteoblast on the outer surface of a bone lay down new bone material, while osteoclast originated from hematopoietic cells break down the old bone for resorption of minerals; the balance between their activities sustains bone mass. A protein called RANKL plays a role in maintaining that balance and, thus, in how bone gets successfully remodeled.
Using osteoblasts and bone marrow cells from mice, plus a synthetic molecule analogous to dsRNA, the study authors experimented with exposure of the cells to dsRNA. They observed that the dsRNA clearly induced the differentiation of more osteoclasts, the cells that break down bone.
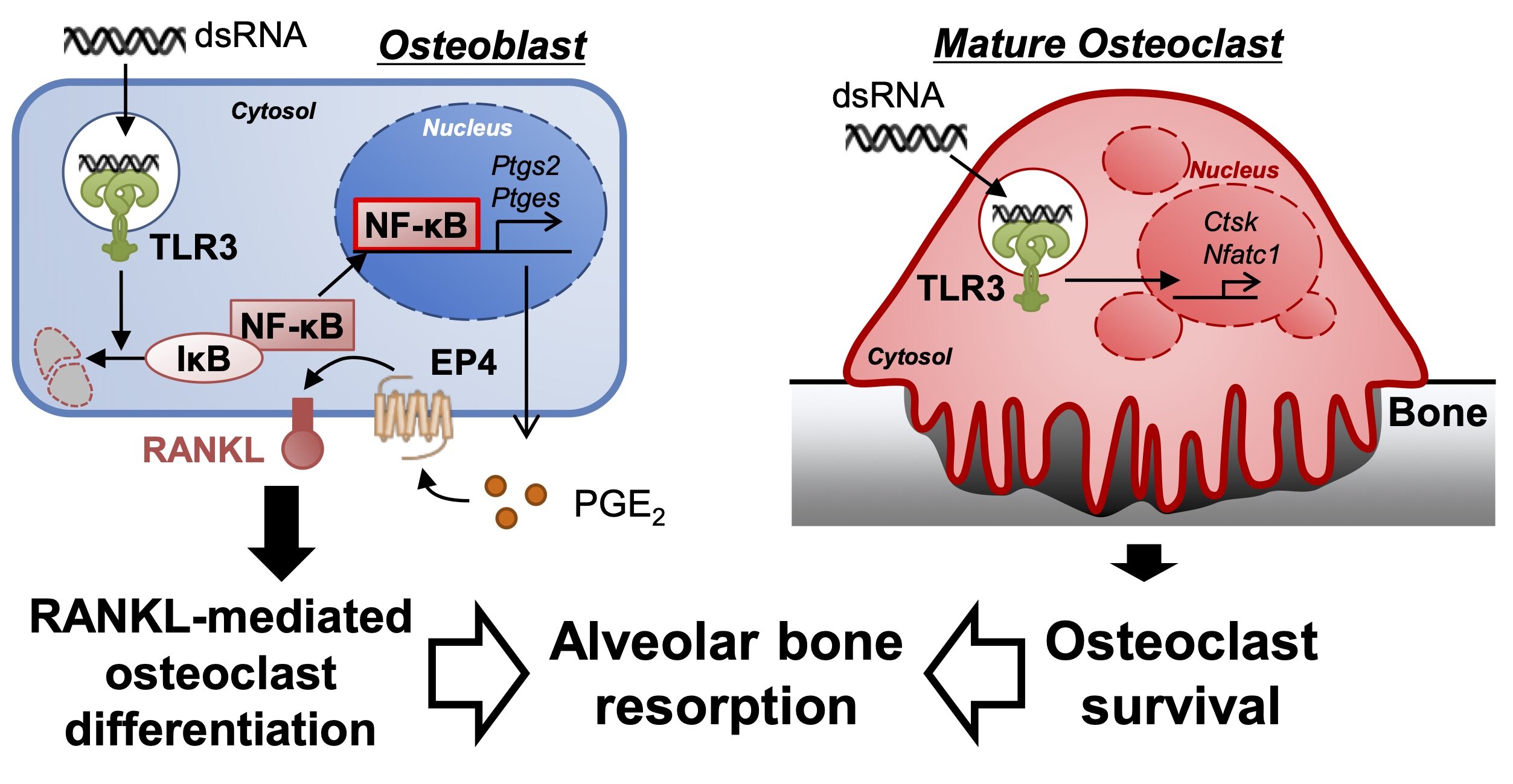
The dsRNA caused osteoblasts to produce more of the hormone-like PGE2 that in turn upregulated RANKL and stimulated osteoclasts to differentiate. So, the osteoblasts, through interactions with the dsRNA molecules, sent cellular signals that increased the production of the bone-eroding osteoclasts. The dsRNA also made mature osteoclasts survive longer.
More, longer-surviving osteoclasts lead to more adsorption of bone when gums are inflamed from bacterial disease. The study revealed a previously unknown mechanism by which gum disease causes breakdown of bones. Says Inada, “These data suggest that TLR3 signaling in stromal osteoblast controls PGE2 production and induces the subsequent differentiation and survival of mature osteoclasts.” The stromal osteoclasts lead to inflammatory resorption of bones anchoring the teeth. Knowing that the inflammation leading to bone damage in periodontitis can be set off by dsRNA introduced via the bacteria or an accumulated immune cells in tissues is a leap forward in combatting the effects of gum disease.
Looking ahead, the researchers plan to further examine how dsRNA – by signaling immune system receptors on stromal osteoblasts to make more PGE2—contributes to progression of periodontitis over time.
Periodontal disease is a local inflammatory disease associated with bacterial infection that results in alveolar bone resorption and tooth loss. The accumulation of bacterial plaque in the periodontal pocket leads to the pathogenesis and progress of periodontal disease.
Lipopolysaccharide (LPS), a membrane component of gram-negative bacteria, is a well-known microbe-associated molecular pattern that causes inflammatory periodontal disease (1). In our previous study, which reported an experimental model of periodontal disease, the production of prostaglandin (PG) E2 mediated severe bone loss (2).
Mice lacking membrane-bound PGE synthase (mPGES)-1, an inducible enzyme for PGE synthesis, failed to develop alveolar bone loss by LPS injection, suggesting that mPGES-1-induced PGE2 synthesis is essential for the LPS-mediated development of periodontal disease.
The balance between osteoclastic bone resorption and osteoblastic bone formation maintains bone mass. For osteoclast differentiation, receptor activator of NF-κB (RANK) ligand (RANKL), which belongs to the tumor necrosis factor superfamily, is an essential molecule.
RANKL expression is induced by several factors, including inflammatory cytokines on the membrane of osteoblasts (3, 4, 5). RANK-expressing hematopoietic macrophages as osteoclast precursor cells interact with RANKL-expressing stromal osteoblasts. RANK-RANKL interaction activates several signaling pathways, such as the MAPK/AP-1 pathway, NF-κB pathway, and calcium pathway, resulting in the activation of nuclear factor of activated T cells, cytoplasmic 1 (NFATc1), a master transcription factor for osteoclast differentiation.
NFATc1 then induces various osteoclast marker proteins, such as cellular fusion factors including osteoclast stimulatory transmembrane protein and dendritic cell-specific transmembrane protein, as well as proteases, cathepsin K and tartrate-resistant acid phosphatase (TRAP).
The osteoclast precursor cells then fuse with each other and differentiate into bone-resorbing osteoclasts (6, 7, 8).
PGE2 is considered a major inflammatory mediator via the upregulation of RANKL in periodontal disease (2). The inflammatory response upregulates the synthesis of the enzymes associated with the production of PGE2, namely, cytosolic phospholipase A2, cyclooxygenase (COX)-2, and mPGES-1.
Arachidonic acids are released from the phospholipid membrane by cytosolic phospholipase A2, then COX-2 converts arachidonic acid into PGH2, and mPGES-1 mediates the synthesis of PGE2 from PGH2. EP receptors (EP1, EP2, EP3, and EP4) are identified as PGE2 receptors.
We have reported that EP2 and EP4 are involved in PGE2-mediated bone resorption and EP4 is reported to be the major receptor associated with PGE2-mediated bone resorption (9, 10, 11). Previous studies have shown that inflammatory molecules, such as LPS and inflammatory interleukins, increased the PGE2 production via the NF-κB pathway.
PGE2 bound to EP4 then enhances the RANKL expression via the activation of CREB transcription factor in osteoblasts, leading to osteoclast differentiation (10, 11, 12, 13, 14).
Toll-like receptors (TLRs) play essential roles in the innate immune response, recognizing various microbial components (15). In mammals, TLR1, 2, 4, 5, and 6 are expressed on the cell surface, whereas TLR3, 7, 8, and 9 localize at the endosomal membrane. LPS, an outer membrane component of gram-negative bacteria, is a potent inducer of periodontal disease.
LPS and TLR4 signaling promotes proinflammatory cytokine production in periodontal tissues (16), stimulates PGE2-mediated RANKL expression in stromal osteoblasts, and inhibits osteogenic differentiation in periodontal ligament stem cells (17), leading to inflammatory alveolar bone resorption.
Synthetic ligands for TLR2/1 and TLR2/6, Pam3CSK4 and Pam2CSK4, were demonstrated to induce PGE2-mediated osteoclast differentiation and alveolar bone destruction in a mouse model of periodontal disease (18). The expression of TLR1 is also reported in periodontal tissues (19, 20) and TLR5, which recognized bacterial flagellin, has been reported to induce osteoclast differentiation and bone loss (21).
These reports indicate that TLRs located on the cell surface are involved in inflammatory alveolar bone loss; however, there is a little evidence of the roles of endosomal TLRs in inflammatory bone resorption, such as periodontal disease. One of the endosomal TLRs, TLR3 in hematopoietic immune cells, recognizes double-stranded RNA (dsRNA) derived from viruses, bacteria, and dead cells (22, 23, 24, 25, 26).
TLR3-dsRNA signaling activates NF-κB and type I interferon signaling, leading to innate immune responses. Recently, polyinosinic-polyinocytidylic acid [poly(I:C)] has been widely used as a TLR3 ligand for the investigation of TLR3 signaling. Poly(I:C) promoted the development of arthritis by inducing the production of proinflammatory cytokines via TLR3 signaling (27, 28); however, it is not clear if TLR3 signaling plays a role in inflammatory diseases, such as periodontal bone resorption in periodontitis.
In the present study, we examined the roles of TLR3 and the ligand in inflammatory bone resorption. Poly(I:C) internalized into the endosome and induced the PGE2-mediated expression of RANKL via the NF-κB signaling pathway in stromal osteoblasts, leading to osteoclast differentiation. Poly(I:C) also prolonged the survival of differentiated mature osteoclasts.
Our data indicate that poly(I:C)-mediated TLR3 signaling in stromal osteoblasts and osteoclasts collaboratively contributes to the development of alveolar bone loss in periodontal disease.
reference link: https://www.jbc.org/article/S0021-9258(22)00043-6/fulltext
More information: Tsukasa Tominari et al, Endosomal TLR3 signaling in stromal osteoblasts induces prostaglandin E2–mediated inflammatory periodontal bone resorption, Journal of Biological Chemistry (2022). DOI: 10.1016/j.jbc.2022.101603



{kind=link}