")










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Viruses infect cells mainly via specific receptors at the cell surface. Antibody-dependent enhancement (ADE) of infection is an alternative mechanism of infection for viruses to infect immune cells that is mediated by antibodies and IgG receptors (FcγRs).
Because ADE of infection contributes to the pathogenesis of some viruses, such as dengue virus and feline coronavirus, it is important to evaluate the precise mechanism of ADE and its contribution to the pathogenesis of SARS-CoV-2. Here, using convalescent-phase plasma from COVID-19 patients, we found that two types of FcγRs, FcγRIIA and FcγRIIIA, mediate ADE of SARS-CoV-2 infection.
Although ADE of infection was observed for SARS-CoV-2 and its recent variants, proinflammatory cytokine production in monocyte-derived macrophages was not upregulated. These observations suggest that SARS-CoV-2 infection produces antibodies that elicit ADE of infection, but these antibodies may not be involved in aberrant cytokine release by macrophages during SARS-CoV-2 infection.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of coronavirus disease 2019 (COVID-19), has spread rapidly around the world and caused a devastating pandemic (1); as of May 2021, there have been more than 166,220,000 cases and 3,445,000 deaths worldwide (2).
The SARS-CoV-2 and its variants continue to ravage human health and the global economy.
During a pandemic, a global vaccination campaign is essential to mitigate the risk of infection and spread (3). To date, several vaccines have been developed and approved (4). However, one of the biggest safety concerns with vaccines is a phenomenon known as antibody-dependent enhancement (ADE) of virus infection (5).
ADE of infection should also be a consideration when patients are being treated with convalescent-phase plasma or monoclonal antibodies (5). Moreover, with the emergence of SARS-CoV-2 variants, the risk for reinfection also raises the possibility of ADE of infection.
ADE is an alternative mechanism of virus infection of cells (5–7). An immune complex of virus and antibodies (mostly nonneutralizing antibodies or cross-reactive antibodies) can bind to receptor molecules, called Fcγ receptors (FcγRs), on immune cells and be internalized, which leads to enhancement of virus entry (5, 7).
Because macrophages/monocytes express FcγRs (FcγRIA, FcγRIIA, and FcγRIIIA) on their surfaces (7–9), macrophages are considered the major inducers of ADE of infection. Moreover, hyperinflammation is often caused by immune cells, including macrophages, upon ADE of various viral infections (10).
ADE of infection occurs with a variety of viruses, including dengue virus, respiratory syncytial virus, and influenza virus, as well as the coronaviruses SARS-CoV-1 and Middle East respiratory syndrome coronavirus (MERS-CoV) (5, 6).
Several studies have been performed to investigate whether SARS-CoV-2 infection induces ADE of infection (11, 12), and ADE of SARS-CoV-2 infection was observed in a study of convalescent-phase-plasma therapy (12).
While FcγRIIA was reported to mediate ADE of SARS-CoV-2 infection in that study, the precise mechanism was not fully elucidated. In addition, it remains unclear whether FcγRIA and FcγRIIIA are involved in ADE of SARS-CoV-2 infection, although they have been reported to mediate ADE of infection with porcine reproductive virus and respiratory syndrome virus (13) and with dengue virus (14) and Japanese encephalitis virus (15), respectively.
To address these unknowns, here, we investigated the mechanism of ADE of SARS-CoV-2 infection by using convalescent-phase plasma from COVID-19 patients and found that ADE of infection is mainly mediated by two types of FcγRs: FcγRIIA and FcγRIIIA.
REFERENCE LINK : https://journals.asm.org/doi/10.1128/mBio.01987-21
Mechanisms of ADE
ADE has been documented to occur through two distinct mechanisms in viral infections: by enhanced antibody-mediated virus uptake into Fc gamma receptor IIa (FcγRIIa)-expressing phagocytic cells leading to increased viral infection and replication, or by excessive antibody Fc-mediated effector functions or immune complex formation causing enhanced inflammation and immunopathology (Fig. 1, Box 1).
Both ADE pathways can occur when non-neutralizing antibodies or antibodies at sub-neutralizing levels bind to viral antigens without blocking or clearing infection. ADE can be measured in several ways, including in vitro assays (which are most common for the first mechanism involving FcγRIIa-mediated enhancement of infection in phagocytes), immunopathology or lung pathology.
ADE via FcγRIIa-mediated endocytosis into phagocytic cells can be observed in vitro and has been extensively studied for macrophage-tropic viruses, including dengue virus in humans16 and FIPV in cats15. In this mechanism, non-neutralizing antibodies bind to the viral surface and traffic virions directly to macrophages, which then internalize the virions and become productively infected.
Since many antibodies against different dengue serotypes are cross-reactive but non-neutralizing, secondary infections with heterologous strains can result in increased viral replication and more severe disease, leading to major safety risks as reported in a recent dengue vaccine trial13,14.
In other vaccine studies, cats immunized against the FIPV S protein or passively infused with anti-FIPV antibodies had lower survival rates when challenged with FIPV compared to control groups17.
Non-neutralizing antibodies, or antibodies at sub-neutralizing levels, enhanced entry into alveolar and peritoneal macrophages18, which were thought to disseminate infection and worsen disease outcome19.
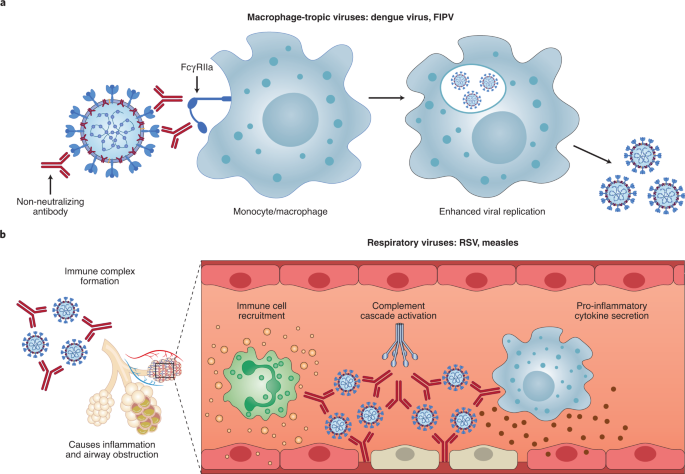
In the second described ADE mechanism that is best exemplified by respiratory pathogens, Fc-mediated antibody effector functions can enhance respiratory disease by initiating a powerful immune cascade that results in observable lung pathology20,21.
Fc-mediated activation of local and circulating innate immune cells such as monocytes, macrophages, neutrophils, dendritic cells and natural killer cells can lead to dysregulated immune activation despite their potential effectiveness at clearing virus-infected cells and debris.
For non-macrophage tropic respiratory viruses such as RSV and measles, non-neutralizing antibodies have been shown to induce ADE and ERD by forming immune complexes that deposit into airway tissues and activate cytokine and complement pathways, resulting in inflammation, airway obstruction and, in severe cases, leading to acute respiratory distress syndrome10,11,22,23.
These prior observations of ADE with RSV and measles have many similarities to known COVID-19 clinical presentations. For example, over-activation of the complement cascade has been shown to contribute to inflammatory lung injury in COVID-19 and SARS24,25.
Two recent studies found that S- and RBD-specific immunoglobulin G (IgG) antibodies in patients with COVID-19 have lower levels of fucosylation within their Fc domains26,27—a phenotype linked to higher affinity for FcγRIIIa, an activating Fc receptor (FcR) that mediates antibody-dependent cellular cytotoxicity.
While this higher affinity can be beneficial in some cases via more vigorous FcγRIIIa-mediated effector functions28,29, non-neutralizing IgG antibodies against dengue virus that were afucosylated were associated with more severe disease outcomes30.
Larsen et al. further show that S-specific IgG in patients with both COVID-19 and acute respiratory distress syndrome had lower levels of fucosylation compared to patients who had asymptomatic or mild infections26. Whether the lower levels of fucosylation of SARS-CoV-2-specific antibodies directly contributed to COVID-19 immunopathology remains to be determined.
Importantly, SARS-CoV-2 has not been shown to productively infect macrophages31,32. Thus, available data suggest that the most probable ADE mechanism relevant to COVID-19 pathology is the formation of antibody–antigen immune complexes that leads to excessive activation of the immune cascade in lung tissue (Fig. 1).
reference link: https://www.nature.com/articles/s41564-020-00789-5



{kind=link}